CASOS CLÍNICS
Malaltia renal poliquística autonòmica recessiva tipus infantil en dos nounats consecutius de la mateixa mare.
Infantile type renal autosomal recessive policystic disease in two successive newborns from the same mother.
Francisco Javier Torres Gómez, Antonio Silva Abad i Glòria Reina Vinardell1.
Serveis d’Anatomia Patològica i de Ginecología1. Hospital de Jerez de la Frontera. Cadis. Espanya.
Direcció per a correspondència
RESUM
Objectiu: La malaltia renal poliquística renal autonòmica recessiva és responsable d’un elevat nombre de defuncions d’aquells que la pateixen ( formes prenatal i neonatal principalment).
Mètodes: Presentem el cas dels estudis necròpsics de dos fills successius de la mateixa mare portadors d’aquesta malaltia.
resultats: Analitzem les característiques macro i microscòpiques d’aquesta entitat quística. Conclusions: Els pacients que aconsegueixen superar la lactància segurament patiran la complicacions derivades de la patologia hepàtica associada.
Paraules clau: Autosòmic recessiva. Malaltia renal poliquística.
SUMMARY
Objectiu: Renal autosomal recessive policystic disease is responsible for a great number of deaths among Affected individuals (mainly prenatal and neonatal forms).
Methods: We report the necropsy studies of two successive newborns from the same mother with such disease.
Results: We analyze the macroscopic and microscopic characteristics of this cystic disease.
Conclusions: Those children that make it through the breast-feeding Període will surely suffer from the associated liver pathology.
Key words: autosomal recessive. Polycystic renal disease.
Introducció
Si bé la malaltia renal autosòmica recessiva és de sobres coneguda, resulta curiós observar la seva presentació en dos fills consecutius de la mateixa mare sobretot avui dia en què les tècniques d’imatge estan prou desenvolupades com per assessorar els progenitors en vista a prendre precoçment l’actitud més adequada a el cas prèvia a la mort de la petita. Aprofitem l’ocasió per il·lustrar els aspectes macro i microscòpics més rellevants d’aquesta patologia.
Cas clínic
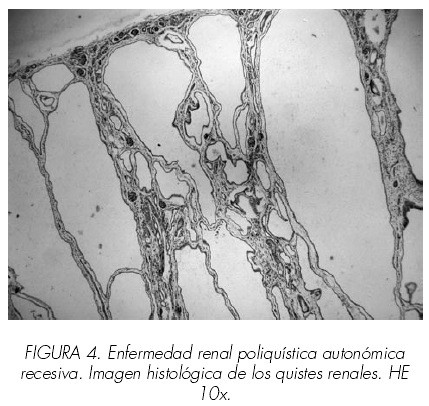
Dos nadons, home i dona d’una mateixa mare i morts als 10 i 45 minuts de vida respectivament als quals se’ls va realitzar examen necròpsic. El primer dels cadàvers, corresponent a la femella, va ser remès amb el judici clínic d’insuficiència respiratòria greu amb sospita de síndrome de Potter amb la constatació de oligoamnios sever; va néixer mitjançant cesària urgent per presentació de natges i el test d’Apgar va ser 1/3/7; minuts més tard va morir. L’examen extern va permetre observar una tonalitat subcianótica, fàcies triangular amb esquerdes parpebrales mongoloides, micrognàtia, arrel nasal ampla i occípit prominent. L’abdomen, globulós, dur i lleugerament abollonado permetia la palpació de dues grans masses ocupant les dues fosses renals i hemiabdomenes. A l’obertura de cavitats destacava la presència de dos grans masses renals de 10 x 8 x 5,5 cm i 12 x 8 x 6 cm amb pesos de 190 i 235 gr respectivament. Si bé es podia destriar la silueta renal, la superfície, abollonada, presentava nombroses formacions quístiques de contingut serós; a el tall aquests quists mostraven una mida heterogeni sent majors els situats a nivell cortical, donant a l’ronyó un aspecte d’esponja. Els pulmons dret i esquerre pesaven 17 i 15 gr (pes habitual del conjunt de 49 gr) mostrant una tonalitat rogenca uniforme; tots dos es trobaven comprimits com a conseqüència de l’elevació diafragmàtica condicionada per la gran grandària dels ronyons. La resta dels òrgans no mostrava alteracions macroscòpiques significatives llevat de les alteracions posicionals derivades de la compressió renal. En el segon dels cadàvers, el corresponent a l’home, es van observar canvis morfològics similars si bé la mida exhibit pels ronyons era encara més gran, amb pesos de 300 i 310 gr. La resta de les vísceres abdominals estaven comprimides contra el diafragma. En tots dos casos es va realitzar un estudi histològic detallat, centrat especialment en els ronyons en els quals es van demostrar múltiples quists de diferents mides amb morfologia sacular a nivell cortical. Aquests quists ocupaven la major part de l’parènquima corticomedular si bé les zones conservades no mostraven alteracions significatives excepte immaduresa focal. Aquests quists estaven entapissats per un epiteli simple que variava des pla o cúbic. Els quists medul·lars, de menor grandària i més arrodonits estaven entapissats per un epiteli de predomini cúbic.Després de les renals, les alteracions més cridaneres es trobaven al fetge on es van observar proliferació i dilatació, fins i tot quística, dels productes biliars a nivell dels espais porta. Amb tals troballes es va emetre en ambdós casos el diagnòstic de malaltia poliquística renal autosòmica recessiva infantil.





Discussió
La malaltia renal poliquística infantil s’ha dividit tradicionalment en dues entitats diferenciades pel que fa a pronòstic i tractament: la autosòmica recessiva, pròpia de nadons i infants i la dominant, que si bé es pot manifestar en les mateixes edats, és característica d’edats més avançades. Les característiques clíniques, radiològiques i macroscòpiques de les dues són similars havent de ser necessari en ocasions recórrer a l’estudi familiar i genètic per consolidar un diagnòstic de certesa, important de cara a l’consell que se li ha de donar a les famílies.
Des del punt de vista morfològic, els ronyons de l’nounat presenten grans dimensions, són no funcionants i mostren quistificació difusa. De manera concomitant el fetge presenta en la majoria de les ocasions una proliferació de conductillos biliars en les àrees porta de fetge. Mentre les manifestacions renals són més importants i rellevants que les hepàtiques en el nounat, aquesta relació es va invertint conforme les manifestacions ocorren a edats més avançades de manera que en adults l’única manifestació pot consistir únicament en fibrosi portal (l’anomenada fibrosi hepàtica congènita ). En el nadó la mida renal pot arribar a ser alarmant comprimint en la majoria dels casos els pulmons, que al seu torn són hipoplásicos i condueixen ràpidament a una situació de distrès respiratori sever, principal causant de la mort en aquests pacients. Al seu torn, la singlot o afunción renal condiciona oligoamnios i en conseqüència compressió facial i fàcies Potter. Per la seva banda, aquells pacients que aconsegueixen superar la infància van a mostrar un predomini de les lesions hepàtiques en forma de fibrosi periportal laxa i proliferació de conductillos biliars, situació que ha estat descrita a la literatura com fibrosi hepàtica congènita, causa d’hipertensió portal i per tant factor predisponerte a el desenvolupament de esplenomegàlia, principalment, i d’altres manifestacions clíniques derivades de la mateixa com ara varius en el tub digestiu, ulceració així com les pròpies de l’possible fallada hepàtica (3,6).
la silueta renal sol estar respectada si bé als talls seriats s’observa un parènquima d’aspecte esponjós causa de la presència de múltiples estructures quístiques de mida petita que a l’estudi microscòpic tenen un aspecte sacular i estan entapissats per epiteli cúbic. Aquests quists es localitzen tant a nivell medul·lar com cortical sent aquests últims més allargats i grans que els medul·lars, més arrodonits. L’estudi amb lectines identifica els quists com originats en els túbuls col·lectors i / o segments tubulars distals. L’arquitectura renal interquística està respectada encara que s’aprecia edema a nivell cortical. En el fetge s’aprecia proliferació i menys freqüentment quistificació de conductes biliars.
Com més tardanes siguin les manifestacions de la malaltia menor serà el nombre de quists i major l’atròfia i els canvis intersticials al ronyó si bé els canvis hepàtics es mantindran més o menys constants a excepció de l’increment en la fibrosi.
han estat diversos els estudis que han intentat tipifificar la genètica d’aquesta entitat aconseguint identificar el gen PKHD1, situat al cromosoma 6p21-23 i responsable de la codificació de la fibrocistina la qual sembla estar relacionada amb la diferenciació dels túbuls col·lectors i els conducte biliars; d’aquí l’alteració de tots dos amb les mutacions de l’esmentat gen (1,2,4,5). Estudis en animals utilitzant aquesta molècula com a diana semblen estar obtenint bons resultats en frenar la progressió de la malaltia.
Bibliografia i lectures recomanades (* lectura d’interès i ** lectura fonamental)
* 1. SWEENEY, W.E .; Avner, E.D .: Molecular and cellular pathophysiology of autosomal polycystic kidney disease (ARPKD). Cell Tissue Res .; 326: 671, 2006.
2. BERGMANN, C .; FRANK, V .; KUPPER, K. i col .: Functional analysis of PKHD1 splicing in autosomal recessive polycystic kidney disease. J. Hum. Genet .; 51: 788, 2006.
3. BASMAISON, O .; LIUTKUS, A .; MICHEL, L. i col .: inherited renal diseases and prenatal diagnosi. Arch Pediat .; 13: 727, 2006.
4. BERGMANN, C .; SENDEREKMJ, Sedlacek i cols .: Spectrum of Mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD / PKHD1). J.Sóc. Soc. Nefrol; 14: 76, 2003.
5. Ward, C.J. Y Cols: “El gen mutat en la malaltia renal poliquística recessiva autosòmica codifica un gran receptor com a proteïna”. Nat. Genet; 30: 259, 2002.
* 6. Kumar, v.; Abbas, A. K. Y Fausto, n.: “Robbins i CORAN Patològica base de la malaltia”; 967. Elsevier, Madrid 2005.