cardioprotateção mediada por canais de potássio dependente do ATP.
Dr Fernando Zeledón SA, Dr. Orlando Morales Ma, Dr. Edgar Méndez JB, Dr. Eduardo Induni LC, Dr. Oswaldo Gutiérrez SD.
Resumo
A mitocôndria desempenha um papel central na manutenção do metabolismo cardiomicito durante os fenômenos da isquemia e reperfusão. Esta “cardiopropeco” parece estar ligada à abertura dos canais de potássio dependentes de ATP na membrana mitocondrial, que evita a abertura do poro de permeabilidade transitória (MPTP), sobrecarga de cálcio e perda de volume de espaço mitocondrial de intermembrana, impedindo a morte celular por necrose ou apoptose .
Vários estudos clínicos sustentam o uso promissor de drogas que abrem esses canais de potássio e que podem ser uma nova arma terapêutica contra a doença isquêmica e suas conseqüências.
palavras-chave: pré-condicionamento isquêmico; canais mitocondriais e sarcolêmicos de potássio; Sobrecarga de cálcio; estresse oxidativo; APOPTOSE, poro de permeabilidade transitória.
Canais de potássio sensíveis a ATPs mitocondriais desempenham um papel importante impedindo a morte celular necrótica e a apopttsis durante os fenômenos de ischamia / reperfusão por meio de impedir por poros de transição de permeabilidade mitocondrial (MPTP) abertura, sobrecarga de cálcio intracelular e perda de espaço intermembrano mitocondrial.
Há evidência clínica de efeitos benéficos de um grupo de bebidas chamadas abridores de canal de potássio que o colud se é uma nova ferramenta terapêutica contra a doença ischânica cardíaca e Sua considência.
Palavras-chave: Precondicionamento isquêmico canais sarcolêmicos e mitocondriais, sobrecarga de cálcio, estresse oxidativo, apoptose, poro transitório de permeabilidade mitocondrial.
introdução
até à data , o tratamento da doença cardíaca da origem isquêmica concentrou-se na prevenção de danos isquêmicos, aumentando a contribuição de oxigênio para a área do miocárdio em perigo ou Diminuindo o consumo de oxigênio de miócito cardíaco.
Da mesma forma, já que há muitos anos, sabe-se que a exposição de miócitos a eventos isquêmicos breves repetidamente, produz proteção contra eventos de isquemia pós-durável, processo chamado de precondicionamento isquêmico (1 ).
Atualmente, o interesse científico é concentrado na busca dos mecanismos celulares envolvidos na cardioproteção contra eventos de isquemia / reperfusão, entre os quais os canais de potássio sensíveis ao ATP (K ATP) parecem ter um papel central (1- 3).
Neste artigo, conceitos de fisiologia celular e molecular relacionados ao papel do ATP K são revisados em defesa isquêmica miocárdica.
Estrutura e função dos canais de potássio cardíaco .
potássio (K +) é a segunda cação mais abundante do organismo. Um adulto de 70 kg contém cerca de 4200 MEQ potássio ou cerca de 50 meq / kg no macho e 40 meq / kg na mulher, levando em conta o ajuste por adipocidade e massa corporal. O conteúdo K + do organismo diminui com a idade em cerca de 2 meq / kg para cada 10 anos diminuindo a massa muscular.
Aproximadamente, 98% de K + está no líquido intracelular, com uma concentração de 150 MEQ / L, enquanto fora das células é de 4 meq / l, com uma restrição de normalidade estreita entre 3,5 a 5,5 meq / l.
A quantidade K + ingerida nos países ocidentais é de quase 50 a 100 MEQ / dia ou mais ou menos 0,7 a 1,3 MEQ / kg de peso corporal por dia. Por exemplo, em uma pessoa que consome 80 meq em um dia, o rim será responsável por excretando 70 MEQ, o trato gastrointestinal 9 MEQ e a pele em torno de 1 MEQ.
potássio está em um saldo estreito , parte do que deve-se a uma redistribuição entre os diferentes compartimentos celulares, se o referido ion é traduzido do meio intracelular para o extracelular ou vice-versa (4). Para ocorrer tal fenômeno, a célula usa proteínas transmembranas que funcionam como canais.
Os canais de íons têm três propriedades essenciais:
1) Um túnel central ou poro através do qual flui de íons em relação ao seu gradiente eletroquímico.
2) Um filtro de seletividade, que determina qual íon será permitido passar pelo poro, chamado região P
3) uma estrutura que exerce a função do portão, controlando a probabilidade conformacional de abertura e fechamento do canal e, portanto, a permeabilidade da referida proteína (4,5,6,7).
Os canais de potássio são compostos de alfa e subunidades beta. Responsável pela condução de íons através do Lipid Bilayer é a subunidade alfa.Dependendo da sua topologia e do número de regiões P (seletividade por poro), foi estabelecida uma classificação de canais de potássio (4,7,8).
Assim, por exemplo, os canais de potássio que aumentam o diminuição Sua permeabilidade dependendo da tensão transmembrana, eles têm seis segmentos transmembranares (TM) e por poro (1 p) e são chamados de canais K + dependentes de tensão (KV, figura 1, A); Aqueles que apresentam 2 TM-1P são canais que permitem a passagem de potássio em relação à intracelular quando a membrana plasmática está em uma tensão mais negativa do que o potencial de equilíbrio e é fechado para potencial mais positivo, então eles são chamados, canais de potássio retificadores para dentro (Kir , K interiormente retificador) (7,9,10,11,12,13).
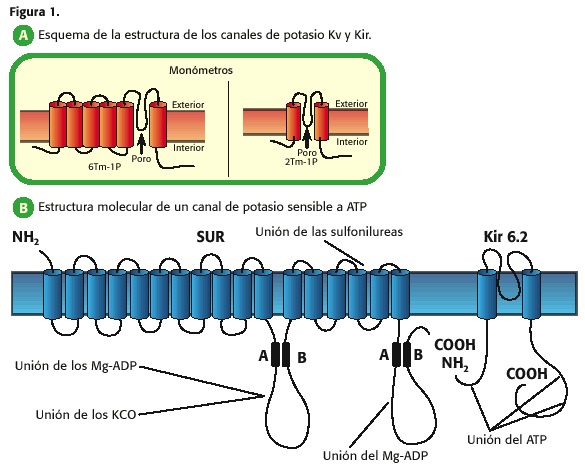
Todas essas famílias de canais de potássio podem, por sua vez, as subfamílias presentes, dependendo da eletrofisiologia do canal ou do ligando que mediam a abertura de tais proteínas, tabela 1.
tabela 1. nomenclatura e estrutura de canais de potássio (14).
dentro dos canais retificadores em internos são duas subfamílias importantes na fisiologia cardíaca: 1) os canais K regulados pela proteína G, que são ativados pelo R. Ecetradores muscarinos m3 (Kach) e 2) canais sensíveis ao ATP (KATP), estes mais recentes objectivos centrais da revisão (7,9,10,13).
A função do ATP K é melhor Conhecida em células pancreáticas beta, onde altera o acoplamento na excitabilidade elétrica da membrana plasmática e a liberação de insulina, em relação à concentração de glicose no sangue. No entanto, eles também foram envolvidos em proteção neural em eventos isquêmicos e epilepsia, regulamento de tom pulmonar e sistêmico), reclinância de glicose no músculo esquelético e proteção contra isquemia de miócito cardíaco, tópico desta revisão (10).
Estas propriedades derivam da capacidade dos canais Katp para acoplar o metabolismo celular à atividade elétrica, detecendo mudanças no citosol de níveis de ATP e adenosina difosfato (MG-ADP), operando o ATP como um bloqueador de canal, e o MG- ADP como um ativador ou promotor de sua abertura (10,11).
O poro dos canais de potássio é um tetrâmer e porque o Kir é 2 TM-1P, quatro dessas unidades estão unidas para formar o Poro, denominado no nível cardíaco Kir 6.2 e Kir 6.1. Além disso, há outra proteína chamada receptor sulfonilurea (sul, receptor sulfonilurea, figura 1, B (15))
que regulamenta a abertura ou fechamento do poro Kir 6.2 ou Kir 6.1: o ATP inibe O canal já se junta ao kir 6 subunit, enquanto o MG-ADP a ativa através de sua interação com a subunidade sul (10,11,12,13,14).
função central das mitocôndrias em Cardioprotecção
mitocôndrias são encontradas em quase todas as células, com exceção de hematis e seu número varia de acordo com o tipo de célula; Por exemplo, cada hepatócito tem 1.022 a 2.000 mitocôndrias, que medem 3 μm de comprimento aproximadamente.
mitocôndrias possuem duas membranas, um externo e outro interno, que resultam em compartimentos de intermembrana e a matriz mitocondrial. Na matriz mitocondrial e na membrana interna, é onde a maioria das atividades relacionadas à cadeia respiratória (16) são desenvolvidas. Nas mitocôndrias, existem várias proteínas que desempenham funções específicas, sejam promovidas cardioproteção ou em detrimento da função cardíaca; e eles são inter-relacionados com o katp por diferentes mecanismos.
Dentro dos mais importantes são:
1) O poro de permeabilidade transicional (MPTP, por poro de transição de permeabilidade mitocondrial) que é composto por:
– canal de anião dependente de tensão (VDAC, canal de anião dependente de tensão), que está localizado na membrana mitocondrial externa (MME)
– o transportador de nucleotídeos de adenina (formiga, ADENINE nucleotide translocator), localizada na membrana interna (MMI)
– a creatina quinase (CK), no espaço enâmbio (EIM)
2) O contra-transportadores Na + / H +, Na + / CA ++, K + / H +, H + / Piruvate, o CA ++ Cotransporter e as proteínas conhecidas da cadeia respiratória, beta-oxidação e ciclo de Krebs (17,18) ( Figura 2).
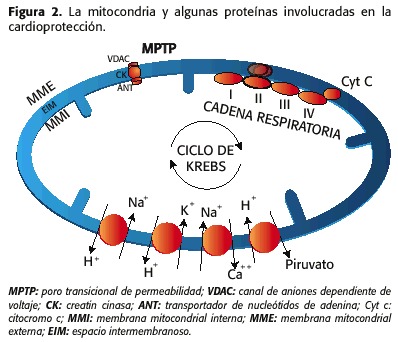
mais importantes envolvidos na gênese da isquemia / repercussão são:
1 Mudanças no volume da matriz mitocondrial
2. O status de eim
3. A permeabilidade do MMI
4. A ruptura do MME e
5. A sobrecondição intramitocrística de cálcio-sódio.
Fisiologia mitocondrial de manuseio de cálcio.
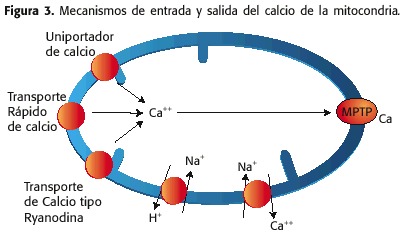
cálcio desempenha um papel fundamental na fisiologia mitocondrial, cujas concentrações estão em faixas estreitas. O cálcio pode ser recapturado para a matriz mitocôndria através do MMI por três mecanismos:
1) Uma bomba de cálcio, conhecida como o Uni-cálcio ou para cima,
2) um canal que permite a entrada rápida de cálcio, conhecida como proteína de transporte de cálcio ou RAM rápida (modo rápido CA ++ Upteke) e
3) um Riverodinia ou Ryr (receptor de ryanodine).
no outro a mão, a saída de cálcio da matriz mitocondrial deve:
1) o contra-transportador de Na + / CA ++, sob condições fisiológicas
2) o MPTP, Sob condições patológicas (Figura 3)
Função primária do cálcio ao nível de A mitocôndria é a estimulação da cadeia respiratória em vários níveis, causando um aumento na produção de ATP e sua exportação para o citosol para atender a demanda metabólica de miocit.
também sabe que o processo de fosforilação oxidativo gera um PE. Quantidade de radicais livres como um produto de complexos I, coenzima Q e complexo III, e que longe de ser prejudicial, é necessário para a transdução de sinal de múltiplos vias metabólicas intramitocriais (19).
mecanismos que promovem Sobrecarga intramitocrística de cálcio. Durante a isquemia miocárdica, o aumento da glicólise causa um acúmulo progressivo de ácido láctico, diminuindo o pH mitocondrial e que exercerá feedback negativo que eventualmente
inibirá a produção glicólise e a ATP. A ativação do Anti-Go + / H + Antecipador que supera as hidrogeniões da mitocôndria busca a normalização do pH intramatricial, mas no processo uma sobrecarga de sódio será encorajada, o que não pode ser bombeado para fora da célula, já que O Na + / K + -atpase é inibido pela perda de ATP (19). Consequentemente, a atividade do NA + / CA ++ contra-transportadora, que geralmente exporta o cálcio das mitocôndrias e da célula, é diminuída ou mesmo atuando inversamente (porque este é um mecanismo de transporte ativo secundário) de modo que nos diferentes compartimentos celulares inicia um feedback positivo que promove grandemente uma sobrecarga de cálcio. Neste ponto, a homeostase iônica não pode ser mantida, as concentrações intracelulares de sódio e cálcio aumentam progressivamente, com diminuição dos nucleótidos de adenina e aumento do fosfato.
Além disso, o aumento do cálcio mitocondrial promove a formação de radicais livres nas mitocôndrias pelos seguintes mecanismos:
1) A sobrecarga do cálcio mitocondrial promove um aumento no fluxo de elétrons na cadeia respiratória
2) também estimula a sintetase nítrica de óxido, formando óxido nítrico (NO) / p>
3) é conhecido que não inibe a cadeia respiratória no nível complexo IV O que
4) aumenta a formação de radicais livres (ROS, espécies reativas de oxigênio) por ciclo Q
5) complexos I e II da cadeia respiratória também podem ser inibidos pelo aumento do cálcio e não, contribuindo ainda mais para a formação de ROS
6) O cálcio também dissocia o citocromo c de proteína cardiolipina em mmi e finalmente MO
7) O aumento do cálcio promove a abertura do MPTP, permitindo a liberação de citocromo, através do MME (19).
Apesar de todos os itens acima, durante a isquemia o mitocondrial PH (PHMI) permanece baixo e este fenômeno, como será discutido nas seções a seguir, é poderoso o suficiente para manter o MPTP fechado, desde que o evento Ischemia não prolongue ou supere a reperfusão temida.
O poro de permeabilidade transitória (MPTP)
O MPTP foi descrito por Haworth e Hunter há mais de 25 anos, no entanto, é até alguns anos atrás que sua função tomou grande importância nos estados de isquemia- reperfusão (20,21,22). Em um estado estacionário ou homeostasia mitocondrial, o MPTP está fechado, a distância
entre MME e MMI é ideal e, portanto, há um acoplamento entre o VDAC-CK-Form, que permite que o mecanismo de energia principal do A mitocôndria é o sistema de creatina-fosfocreatina e, em segundo lugar, do ATP / ADP (Figura 4) (23). Além disso, a permeabilidade do ANT-VDAC é baixa neste estado e os canais de potássio dependentes ATP estão fechados (18,23,24).

Por outro lado, o antipersporter k + / h + leva potássio de A matriz em troca de uma hidrogenión, e também tem uma baixa cinética em condições estacionárias, para garantir a manutenção de fosforilação oxidativa, volume de matriz adequado e um potencial de membrana mitocondrial negativo, uma vez que é conhecido, o potencial das hidrogenões entre o EIM e a A matriz mitocondrial é mantida na impermeabilidade do MMI para os diferentes íons, que permite a geração ATP pelo complexo V localizado no MMI.
Em conclusão, de um ponto de vista fisiológica, o regulamento Do volume da matriz mitocondrial tem consequências importantes para o metabolismo energético de miocus e o estado impermeável do pore contribui para a referida homeostasia (17,24.25).
O poro de permeabilidade transitória durante a isquemia e durante a repercussão . Em condições de isquemia miocárdica, o MPTP se abre em menor quantidade, permitindo que o MMI seja permeável a qualquer molécula < 1,5 kDa. Duas conseqüências importantes ocorrem então. Primeiro, proteínas intramitriais não podem passar pelo poro e exercer pressão coloitosmótica no inchaço da matriz. O MMI por causa de sua consistência não sofre lise, no entanto, tal fenômeno ocorre em MME, com a liberação de proteína para EIM, como citocromo C e o fator indutor de apoptose que desempenha um papel crítico na morte celular por esse mecanismo. . Em segundo lugar, o MMI torna-se permeável aos prótons, que desacoplaram a cadeia respiratória, diminuiu a produção de ATP e terceiro, o funcionamento reverso da sintetase ATP é promovido, isto é, em vez de sintetizar ATP é promovido a hidrólise, por causa da manutenção de um gradiente de H + e um potencial de membrana mitocondrial negativo (18,26,28,28,29).
No entanto, em tal situação, as concentrações de ATP recusam rapidamente, dirigindo em uma alteração iônica de homeostase metabólica e a ativação de enzimas que promovem a degradação, como fosfolipases, nucleases e proteases. A menos que o fechamento do MPTP ocorra, essas alterações causam danos celulares irreversíveis, resultando em necrose celular.
Um fator chave na abertura do MPTP é a sobrecarga de cálcio intrraatricial, especialmente quando é acompanhada de estresse oxidativo, depleção de Nucleótidos de adenosina, altas concentrações de fosfato iônico e despolarização do MMI, microambiente que é apresentado em isquemia. Portanto, a abertura do MPTP é um passo crítico na transição de danos por células reversíveis para irreversível (18,23).
Como mencionado acima, o MPTP é formado pelo Ant-VDAC e no outro Proteína, que é chamada de ciclofilina-d (CYP-D), que tem que se juntar à formiga para causar a sua abertura, união promovida pelo aumento da concentração de cálcio, pelo esgotamento das reservas de nucleotídicas de adenosina, aumentando os PHM e as alterações de volume de matriz. No entanto, um poderoso inibidor de abertura de poros é a redução do PHMI, um fenômeno que ocorre durante a isquemia, estabelecendo um equilíbrio entre os fatores que favorecem a sua abertura (sobrecarga mitocondrial de cálcio, depleção ATP, aumento do estresse oxidativo) e aqueles que o mantêm fechados ( sob PHMI), Figura 5.
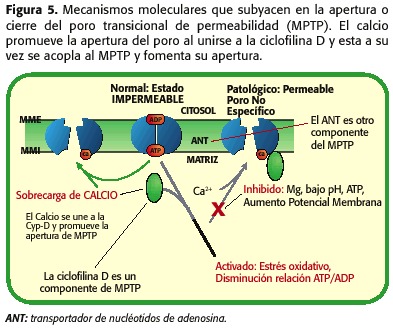
Tão ambos, durante a isquemia o MPTP Abre, mas a baixa quantidade, uma vez que a micro-nação celular não é o ideal para ocorrer tal fenômeno. Por outro lado, durante a reperfusão, há uma série de fenômenos intramitriais que promovem a abertura do MPTP ainda mais do que durante a isquemia. Quando a reperfusão ocorre, a mitocôndria é novamente capaz de respirar e gerar um potencial de membrana que permita a síntese do ATP, com o aumento da produção de radicais livres, como produtos da cadeia respiratória, o PHMI começa a aumentar e ainda persistir Sobrecarga do cálcio e a deflexão do ATP. Portanto, neste momento, promoveu a abertura do MPTP (30,31,32,33,34,35) (Figura 6). Dependendo da proporção de poros abertos e o tempo restante em tal estado, necrose ou apoptose (18,23,24) irá para promover (Figura 7).
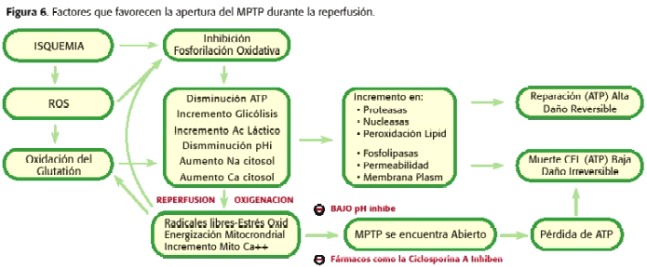
Desta forma, a mitocôndria se torna um objectivo terapêutico, que promove a inibição direta do MPTP ou indirectamente, impedindo que os fenómenos que promovam a sua abertura, tal como / p>
1) a sobrecarga de cálcio que pode ser obtida com o propofol36 ou com o aumento das concentrações de mg ++ que inibe o NA + / CA ++ do transportador de cálcio e o tipo L (37)
2) Melhorar a bioenergética mitocondrial ou o volume da matriz, ambos os fenômenos propiciados com drogas que abrem canais de potássio (KCO, abridores de canal de potássio) vêem abaixo
3) mantendo um baixo pH durante a reperfusão, Por exemplo, inibindo o antipersportador na + / h + com medicamentos como amiloreto, ou adicionando piruvato (38) às mitocôndrias que incentiva o aumento do ácido láctico.
Tudo para Nterta abre uma imensa porta na pesquisa em ciências básicas e em terapêutica para pacientes que sofrem de isquemia miocárdica ou suas conseqüências.
potássio canais sensíveis ao ATP: sua função na prevenção da abertura do Mitocondrial e Na membrana sarcoplasmática
durante situações de estresse miocárdico, em que o ATP é REFT, os canais de K + aumentam seu status de abertura com consequências benéficas para o microambiente de micoCIT (39,40, 41). / P>
A abertura do Mitocondrial K ATP provoca um aumento modesto na influência de K + em relação ao interior da matriz, o que causa dois efeitos diferentes, dependendo do estado bioenergético de cardiomiócito. Primeiro, quando a célula está em repouso, o potencial de membrana mitocondrial é alto, e a entrada de íons de potássio promove a tartaruga mitocondrial e a alcalização da matriz, que estimula um ligeiro aumento na produção de radicais livres. Em segundo lugar, se a célula estiver no estado da isquemia, a mitocôndria é implantada (potencial de baixo potencial), que promove a saída de K +, um fenômeno que é neutralizado pelos canais de K ATP e, portanto, a contração do volume da motrix que caso contrário ocorrer (42).
O rendimento de potássio para a matriz mitocondrial gera uma ligeira produção de radicais livres (ROS), e como é demonstrado, elas desempenham um papel importante como segundo mensageiros em uma variedade de sinais intracelulares (39). Esta produção de ROS em uma célula de repouso também promove a recaptura de K + em troca por um H + (antitransporter k + / h +), que cria o gradiente para a troca de um fosfato (PI) pela eletrônica Cotransporter PI / H + . A recuperação do PI é muito menor que a de K +, porque o PI está presente em muito menos concentração do que o K +. Por esta razão, o pH da matriz é sempre aumentado quando o volume da matriz também aumenta, fenômenos promovidos pelo PI e K + Recapture (23).
Quando os canais ainda não entraram em função do Katp durante eventos isquêmicos, a matriz mitocondrial sofre uma contração e a EIM aumenta, com a desunião do VDAC da formiga pelo CK, o que aumenta a condutância para o trocador de nucleótidos pelo VDAC e a formiga, contrária ao que é Acontecendo em repouso, contribuindo assim para a deflexão da matriz e da ATP citosólica (Figura 3). Portanto, a abertura do Katp durante a isquemia, por exemplo, promovida pelo diaboxido, mantém o volume do EIM, reduz a taxa de perda de ATP, reduz a taxa de degradação dos nucleótidos da adenina de forma que existam reservas de ADP para a subsequente fosforilação durante a reperfusão e, finalmente, reduz as alterações no potencial de membrana mitocondrial e o acúmulo de CA ++, evitando a sobrecarga de cálcio, uma vez que o ATP permanece em concentrações adequadas para operação mínima de Na + / K + ATPASA e outras bombas.
Esses efeitos preservam a função mitocondrial e, dessa forma, podem ser enfrentadas com reperfusão com melhores resultados fisiopatológicos. Durante a reperfusão, a abertura subsequente dos canais KATP permite a compartimentalização dos nucleótidos de adenina, o que significa que a primeira fonte de energia é fosfato de fosfocreatina e não o ATP (baixa condutância do VDAC (23), Figura 3).
Por conseguinte, a activação dos canais ATP mitocondriais durante a isquemia-reperfusão impede a formação do poro de permeabilidade transitória, inibindo o estresse oxidativo, desencadeado pela renda do CA ++ (18,24).
Vanden Hoek et al. Eles mostraram que o aumento de radicais livres na mitocôndria durante a pré-condição isquêmica era necessário para proteger o miocus contra um estresse oxidativo subsequente durante a reperfusão.É assim que tem hipotetizado que existem duas etapas na libertação de radicais livres, a primeira que contribui para a abertura dos canais de potássio sensível ao ATP e subsequente durante a repercussão, que é prejudicial e desencadeia a apoptose remodelada e cardíaca ( 42). Na membrana sarcolêmica do miócito cardíaco e do músculo liso, a abertura de canais de potássio também promove efeitos de proteção.
hiperpolarização celular que impede a sobrecarga intracelular do CA ++, diminuindo a duração do potencial de ação, limitando celular danificar e preservar as reservas de energia celular e, portanto, a sobrevivência do miocit. Além disso, esta sequência pode ser uma figura aguda e crônica (8,42,44). No nível vascular, a abertura desses canais promove vasodilatação, devido à hiperpolarização celular e redução na entrada de CA ++, com aumento coronariano e diminuição da carga.
Perspectivas clínicas
> Devido às propriedades combinadas da cardioproteção e vasodilatação, os medicamentos que permitem a abertura de canais de potássio (KCO), podem ser considerados para certas condições cardíacas (Tabela 2). Estes incluem proteção miocárdica sob circulação extracorpórea, preservação do coração doador no transplante cardíaco, tratamento da doença isquêmica cardíaca, de hipertensão sistêmica e pulmonar, doença vascular periférica e arritmias relacionadas à repolarização anormal (41-50).
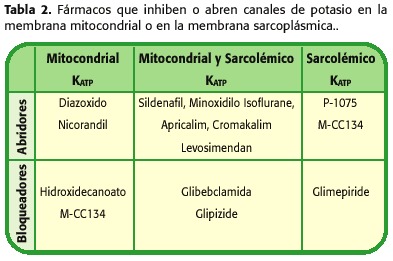
na cirurgia cardíaca, a KCO poderia desempenhar um papel importante para soluções de cardioplegia. Em vários modelos de cirurgia cardíaca com circulação extracorpórea, vários KOC, incluindo Nicorandil, Aprikalim e Pinacidil, promoveram maior cardioproteção do que com cardioplexia convencional (51,52).
em pacientes que foram realizados em cirurgia de pontes (bypass) O tempo necessário para atingir a parada cardíaca, alterações no segmento ST após a fixação da aórtica, os níveis plasmáticos da CPK-MB e dose de agentes inotrópicos, foram todos menores do que o grupo tratado com Nicorandil em comparação com os controles tratados com terapia convencional ( 53).
O uso de soluções cardiopléficas com KCO, como diazoxido, minoxidil (54), propofol (que inibem sobrecarga de cálcio ++ (36)), magnésio (que inibe a sobrecarga de cálcio37), promover cardioprotecção por vias de sinalização de células que convergem com as de KCO (23,55)), piruvato (que encoraja acidose intracelular e Padrão de grau de pH pelo fechamento MPTP (38)), os inibidores do contra-transportadoras Na + / H ++, como a amiloreto, (que também evitam a perda do baixo efeito protetor da baixa), adenosina (que também é um KCO agindo através de proteínas G, impede a sobrecarga de cálcio, uma vez que a cardioplegia hiperpotêmica promove (52,56)), pode ser objetivos fortes para a preservação da função cardíaca em cirurgias de alto risco e pode evitar complicações potencialmente mortais, como a baixa síndrome de gastos pós-bomba e muitos Outros, com redução de custos econômicos, reduzindo as estadias hospitalares.
Por outro lado, o uso de antioxidantes parecem ser benéficos apenas em determinadas circunstâncias, já que seu uso em ensaios laboratoriais bloqueou o pré-revestimento isquêmico porque os radicais livres são liberados em menor quantidade (23,25).
na síndrome coronariana aguda, valor Do KCO é melhor documentado clinicamente com Nicorandil que provou ser benéfico com efeitos adversos mínimos em lidar com angina estável e instável (57). Em um estudo multicêntrico envolvendo mais de 5.000 pacientes com angor estável, o uso a longo prazo de Nicorandil estava associado à redução de eventos cardiovasculares, como morte cardíaca, infarto do miocárdio e hospitalização devido à dor no peito (58.59).
em Pacientes com raiva instável, o Nicorandil adicionou a um tratamento anti-isquêmico agressivo reduziu episódios de isquemia / necrose miocárdica e arritmias quando comparado com os pacientes tratados com terapia convencional. Em pacientes submetidos à angioplastia coronariana, o Nicorandil predominou o coração, melhorou a hemodinâmica coronariana e a viabilidade miocárdica preservada antes da reperfusão (60.61,62).
Nicorandil também diminui a pré-carga e a carga, aumenta a liberação de óxido por células endoteliais e, ao contrário da nitroglicerina, não desenvolve tolerância para seus efeitos antierginosos (63).
Na angina vasaspastal, Nicorandil é um poderoso vasodilatador e demonstrou diminuir os episódios de angina variante, diminui as alterações do segmento ST e melhora a perfusão coronariana. O KOC também demonstraram resultados melhores na cirurgia de bypass coronariano usando enxertos arteriais (artérias torácicas internas, gastroepiplóica, radial), pois evita o vasoespasmo subseqüente (64,65,66)). A experiência e os dados científicos são necessários com o uso de KCO nessas situações clínicas.
A ativação do katp sarcolêmico é responsável pela corrente elétrica sublinhando a elevação do segmento ST, o clássico indicador eletrocardiográfico de danos isquêmicos transmurais miocárdicos ( 41). Pacientes com diabetes mellitus tratados com sulfoniluréias e que são infarto agudo para o miocárdio, apresentam uma magnitude atenuada na elevação de ST, o que torna difícil para o diagnóstico inicial, uma vez que sulfoniluréus são inibidores de canais de katp dependentes (41).
Conclusão
Como praticamente todas as doenças humanas, a terapêutica da doença isquêmica cardíaca baseia-se em novos conhecimentos em fisiologia celular e fisiopatologia. A modulação de canais KATP é um processo crítico na homeostase metabólica da célula e de acordo com a pesquisa biomédica dilui novos conceitos sobre a estrutura, função, regulação e seletividade de tecidos desses canais, novos agentes terapêuticos podem ser desenvolvidos com enormes vantagens para a população em risco.
Referências
1. Ardehali H, O’Rourke. Canais katp mitocondrial na sobrevivência e morte celular. J Mol Cell Cardiol 2005; 39: 7-16.
2. Kloner Ra, Bolli R, Marban e, Reinilib l, Braunwald E. Implicações Médicas e Celulares de deslumbrante, hibernação e pré-condicionamento: um workshop NHLBI. Circulação 1998; 97: 18: 48-67.
3. Murray CE, Jennings RB, Reiner Ka. Precondicionamento com isquemia: um atraso da lesão celular letal no miocárdio isquêmico. Circulação 1986; 74: 11: 24-36.
4. Cogan mg. Líquidos e eletrólitos: fisiologia e fisiopatologia. Manual moderno. México, 1993; 145-168.
5. Sansom MSP, Shrivastava Ih E em. Canais de potássio: estruturas, modelos, simulações. Biochimica et Biophysica Acta 2002, 1565: 294-307.
6. Biggin PC, Roosild T, Choe S. Potássio Estrutura de canal: domínio por domínio. Opinião atual Biologia Estrutural 2000, 10: 456-461.
7. Giblin JP, Leansey JL, e Tinker A. A montagem molecular de canais de potássio sensíveis a ATP: determinantes no pare formando subunidade. J Biol Chem 1999, 274: 22652-22659.
8. Choe S. Estruturas de Canal de Potássio. NAT Rev Neurociência 2002, 3: 115-121.
9. Lousoun G, Rose T, Nichols CG. Base estrutural da gatinha do canal de potássio rectificante interno. Tendências Cardiovasc Med 2002; 12: 253-258
10. Nishida M, Mackinnon R. Estrutural base da retificação interna: poro citoplasmático do retificador de proteína G Girk1 em 1,8 à resolução. Célula 2002; 111: 957-965.
11. Yamada M, Inanobe A, Kurachi Y. G Regulação de proteína de canais de íons de potássio. Avaliações farmacológicas 1998; 50: 724-747.
12. Bichet D, Haass F, Yeh Jan L. Mescando Estudos Funcionais com Estruturas de Retificador Interior K + Canais. Revisões da Nature 2003; 4: 957-67.
13. Aguilar-Bryan L, Clement IV J, González G, Kunjilwar K, Babenko A, Bryan J. para entender a montagem e a estrutura dos canais Katp. Avaliações fisiológicas 1998; 78: 227-242.
14. Elizari M, Chiale P. Arritmias cardíacas: Fundamentos celulares e moleculares, diagnóstico e tratamento. Panamericana, Buenos Aires, 2003; 31-40.
15. Campbell JD, Sanson MSP, Ashcroft F. Potassium Channel Regulation. Revisões da Nature 2003; 11: 1038-1042.
16. Méndez Je, Zeledón SF, Zamora JF, Cortés VA. Uma abordagem para a parte da cinética de oxigênio 1. Rev CostaR Caridiol 2004; 6: 27-32.
17. Suleiman M, Haletrap AP, Griffiths E.J. Mitocôndria: um alvo para proteção miocárdica. Farmacologia e Terapêutica 2001; 89: 29-46.
18. Haletrap AP, Clarke J, Sabzali A, Javadov A. A transição de permeabilidade mitocondrial pare a abertura durante a reperfusão miocárdica: um alvo para cardioproteção. Cardiovasc 2004; 61: 372-385.
19. Brookes P, Yoon e, RoboTham J et al. Cálcio, ATP e ROS: um triângulo mitocondrial do amor-ódio. Sou J Physiol Cell Physiol 2004; 287: C817-C833.
20. Caçador Dr e Haworth Ra. A transição de membrana induzida por CA ++ na mitocôndria I. Os mecanismos de proteção. Biophys biochem de arco 1979; 195: 453-459.
21. Caçador Dr e Haworth Ra. A transição de membrana induzida por CA ++ na mitocôndria II. Natureza do site de gatilho do CA ++. Biophys biochem de arco 1979; 195: 460-67.
22. Caçador Dr e Haworth Ra. A transição de membrana induzida por CA ++ na Mitocôndria III.CA Transitional CA ++ relase. Biophys biochem de arco 1979; 195: 468- 77.
23. Garlid KD, Dos Santos P, Xie ZJ, Costa A, Paucek P. Mitocondrial Potássio Transporte: o papel do canal k + sensível a ATP mitocondrial na função cardíaca e cardioproteção. Biochem biophys acta 2003; 1606: 1-21.
24. Macfallas E, Liem D et al. Função mitocondrial: o coração da preservação do miocárdio. J lab clin med 2003; 142: 141-9.
25. Facundo H, Fornazari M, Kowalsowski A. Proteção tecidual mediada por canais mitocondriais K +. Biochem Biophys Acta 2005; 1701: 1-11.
26. Halestrap ap. A transição de permeabilidade mitocondrial: seu mecanismo molecular e papel na lesão de reperfusão. Em: Brown GC, Nicholls DG, Cooper CE, editores. Mitocôndrias e morte celular. Sociedade Bioquímica Simpósia. Londres: Portland Press; 1999, vol. 66, p. 181-201.
27. Halestrap AP, McStay GP, Clarke SJ. O complexo de poros de transição de permeabilidade: outra visão. Biochimie 2002; 84: 153-66.
28. Halestrap AP, Kerr PM, Javadov S, Woodfield Ky. Elucidando o mecanismo molecular da transição de permeabilidade por poro e seu papel na lesão de reperfusão do coração. Biophim Biophys Acta 1998; 1366: 79-94.
29. Halestrap AP, Brenner C. O nucleotide de Adenine Translocase: um componente central da transição de permeabilidade mitocondrial por poro e player chave na morte celular. Curr Med Chem 2003; 10: 1507-25.
30. Duchen Mr, McGuinness O, Brown La, Crompton M. Sobre o envolvimento de uma ciclosporina – um poro mitocondrial sensível na lesão do reperfusão miocárdica. Cardiovasc res 1993; 27: 1790-4.
31. Lemasters JJ, Nieminen Al, Qian T, Trost LC, Herman B. A transição de permeabilidade mitocondrial na lesão tóxica, hipóxica e reperfusão. Mol Cell Biochem 1997; 174: 159-65.
32. Lemasters JJ, Trollinger Dr, Qian T, Cascio Nós, Ohata H. Imagem confocal de Ca2 +, pH, potencial elétrico e permeabilidade de membrana em células vivas únicas. Métodos Enzymol 1999; 302: 341-58.
33. Xu mf, Wang Yg, Hirai K, Ayub A, Ashraf A. Pré-condições do cálcio inibe a transição de permeabilidade mitocondrial e a apoptose. Sou J Physiol 2001; 280: H899- 908.
34. Miyata H, Lakatta, por exemplo, Stern MD, Silverman HS. Relação de cálcio livre mitocondrial e citosólico para a recuperação do miocito cardíaco após a exposição à anóxia. Circ res 1992; 71: 605-13.
35. Griffiths EJ, Ocampo CJ, Savage JS, et al. Caminhos de transporte de cálcio mitocondrial durante hipóxia e reoxigenação em cardiomiocitos de rato único. Cardiovasc res 1998; 39: 423-33.
36. Lim Khh, Modi P, Nicholson E, et al. Um modelo de porco de parada cardioplégico de sangue quente para investigar os efeitos cardioprotetoras do propofol. J fisiol 2001; 536p: 82p.
37. Headrick JP, McKirdy JC, Willis RJ. Efeitos funcionais e metabólicos do magnésio extracelular no miocárdio normoxico e isquêmico. Sou J Physiol 1998; 275: H917-29.
38. Mallet Rt. Piruvato: Protetor metabólico do desempenho cardíaco. Proc SoC Exp Biol Med 2000; 223: 136- 48.
39. Oldenburg O, Cohen M et al. Canais katp mitocondriais: papel na cardioproteção. Cardiovasc res 55: 429-437, 2002.
40. Minners J, McLeod C, Saco N. Mitocondrial Plasticidade no clássico pré-condicionamento isquêmico, movendo-se além do canal katp mitocondrial. Cardiovas res 2003; 59: 1-6.
41. Kane G, Liu X, Yamada S, Olson T, Terzic A. Canais Cardíacos Katp em Saúde e Doença. J Mol Cell Cardiol 2005; 38: 937-943.
42. Wang Y, Haider H, Ahmad N, Aschraf M. Mecanismos por Wich Katp Channel Openers produzem cardioproteção aguda e atrasada. Farmacologia Vascular 2005; 42: 253-264.
43. VANDEN Hook T, Becker L.B, Shao Z, Schumacker P. As espécies reativas de oxigênio reativas liberadas de mitocôndrias durante a bela hipóxia induzem pré-condicionamento nos cardiomiócitos. J biol. Chem. 1998; 273: 18092-98
44. Jahangir A, Terzic A. Katp Channel Terapeutics no leito. J Mol Cell Cardiol 2005; 39: 99-112.
45. Kane GC, Behfar A, Yamada S, Perez-Terzic C, O’Cochlain F, et al. Hoenicke em, Sun Xw, estranha RG, Damiano RJ. Preservação do coração doador com uma nova solução hiperpolarizadora: proteção superior em comparação com a solução da Universidade de Wisconsin. J thorac cardiovasc surg 2000; 120: 746-54.
46. Quast u, guillon jm, Cavero I. Farmacologia celular de abridores de canal de potássio em músculo liso vascular. Cardiovasc res 1994; 28: 805-10.
47. Okada Y, Yanagisawa T, Taira N. BRL 38227 (Levcromakalim) A hiperpolarização girada reduz a sensibilidade a Ca2 + de elementos contráteis na artéria coronariana canina. Naunyn Schmiedebergs Arch Pharmacol 1993; 347: 438-44.
48. Donnelly R, Elliott HL, Meredith Pa, Reid JL. Estudos clínicos com o ativador do canal de potássio Cromakalim em indivíduos normotensos e hipertensos. J cardiovasc farmacol 1990; 16: 790-5.
49. Simpson D, Wellington K.Nicorandil: Uma revisão do seu uso na gestão de angina estável peitoris, incluindo pacientes de alto risco. Drogas 2004; 64: 1941-55.
50. McCully JD, Levitsky S. Mitocondrial ATP-sensíveis canais de potássio em cardioprotecção cirúrgica. Biophys biochem de arco 2003; 420: 237-45.
51. Kevelaitis E, OUBenaissa A, Peynet J, Mouas C, Menasche P. Pré-condições pré-condicionamento por aberturas de canal de potássio sensíveis a Mitocondriais – Uma abordagem eficaz para melhorar a preservação dos transplantes cardíacos. Circulação 1999; 100: 345- 50.
52. STEENSRUD T, Nordhaug d, Husnes KV, Aghajani E, Sorlie DG. Substituir o potássio por Nicorandil no Cardioplegia Hospital de St. Thomas frio melhora a preservação de energética e função nos corações de porco. Ann Torac Surg 2004; 77: 1391-7.
53. Hayashi Y, Sawa Y, Ohtake S, Nishimura M, Ichikawa H, Matsuda H Controlled Administração de Nicorandil Proteção FormyOCARDIAL Durante a artéria coronariana Bypass enxertia sob o bypass cardiopulmonar. J cardiovasc farmacol 2001; 38: 21-8.
54. Garlid PD, pucou p et al. Efeito cardioprotetivo do diaxóido e sua interação com canais K + sensíveis a ATP mitocondriais: mecanismo ou cardioproteção possível, Circ Res 1997; 81: 1072-182.
55. A interação XIC Z. Ouabain com Cardiac Na / K- ATPASA revela que a enzima pode atuar como uma bomba e como transdutor de sinal. Cell Mol Biol 2001; 47: 383-390.
56. Jovanovic A, López J, Alekseev A et al. A adenosina impede o carregamento CA21 induzido K1: insight sobre cardioproteção durante a cardioplégia. Ann Torac Surg 1998; 65: 586-91.
57. Simpson D, Wellington K. Nicorandil: Uma revisão de sua utilização na gestão de angina estável pectoris, incluindo pacientes de alto risco. Drogas 2004; 64: 1941-55.
58. Markham A, Plosker GL, Goa Kl. Nicorandil – uma revisão atualizada de seu uso em doenças cardíacas isquêmicas com ênfase em seus efeitos cardioprotetoras. Drogas 2000; 60: 955-74.
59. O grupo de estudo de Iona. Julgamento para mostrar o impacto de Nicorandil em Angina (Iona): Design, Metodologia e Gestão. Coração 2001; 85: E9.
60. Ito H, Taniyama Y, Iwakura K, Nishikawa N, Masuyama T, et al. A Nicorandil intravenosa pode preservar a integridade microvascular e a viabilidade miocárdica em pacientes com infarto miocárdico de parede anterior reperfado. J am colar cardiol 1999; 33: 654-60.
61. Schlepper M, Thormann J, Berwing K, Strasser R, Mitrovicv. Efeitos do Nicorandil na perfusão regional e à função ventricular esquerda. Drogas cardiovascas 155; 9: 203-11.
62. Sakatay, Kodama K, Komamura K, Limyj, Ishikura F, et al. Efeito salutar da administração intracoronária adjunta de Nicorandil na restauração do fluxo sanguíneo miocárdico e melhora funcional em pacientes com infarto agudo do miocárdio.AM Coração J 1997; 133: 616-21.
63. Simpson D, Wellington K. Nicorandil: Uma revisão de sua utilização na gestão de angina estável pectoris, incluindo pacientes de alto risco. Drogas 2004; 64: 1941-55.
64. Kaski jc. Gestão de angina vasofástica-papel de Nicorandil. Drogas cardiovascas 155; 9: 221-7.
65. Chen JW, Lee Wl, Hsu NW, Lin SJ, Ting CT, Wang SP, et al. Efeitos do tratamento de curto prazo de Nicorandil na isquemia miocárdica induzida pelo exercício e atividade autonômica cardíaca anormal na angina microvascular. Sou J cardiol 1997; 80: 32-8.
66. Akar F, Uydes-Dogan BS, Tufan H, Aslamaci S, Koksoy C, Kanzik I. A comparação da capacidade de resposta das artérias mamárias internas isoladas humanas e gastroepiplóicas para levcromakalim: uma abordagem alternativa para a gestão do espasmo do enxerto. BR J Clin farmacol 1997; 44: 49-56.
Um departamento de Fisiología, Universidad de Ciencias Médicas (Ucimed), Sabana Oeste, San José, Costa Rica. Teléfono (506) 296-3944, E-mail: [email protected], [email protected]
b cárugía, hospital México, Ucimed, San José, Costa Rica.
C Cádra de Cirugía, Hospital México, Universidad de Costa Rica (UCR), San José, Costa Rica.
D Cádra de Fisiopatología, Hospital México, Ucimed y ucr, San José, Costa Rica