Cardioprotection médiée par des canaux de potassium dépendant de l’ATP.
Dr Fernando Zeledón SA, Dr Orlando Morales Ma, Dr Edgar Méndez JB, DR Eduardo Induni LC, Dr Oswaldo Gutiérrez SD.
Résumé
Le mitochondria joue un rôle central dans le maintien du métabolisme cardiomicito au cours des phénomènes d’ischémie et de reperfusion. Cette « cardioprotection » semble être liée à l’ouverture des canaux de potassium dépendants de l’ATP dans la membrane mitochondriale, ce qui évite d’ouvrir le pore de perméabilité transitoire (MPTP), la surcharge de calcium et la perte de volume d’espace intermembranaire mitochondrial, empêchant la mort cellulaire par nécrose ou apoptose .
Diverses études cliniques maintiennent l’utilisation prometteuse des médicaments qui ouvrent ces canaux de potassium et qui pourraient être une nouvelle arme thérapeutique contre la maladie ischémique et ses conséquences.
Mots-clés: préconditionnement ischémique; canaux de potassium mitochondriaux et sarcolémiques; Surcharge de calcium; stress oxydatif; Apoptose, pore de perméabilité transitoire.
Résumé
Mitochondrial Sensible aux canaux de potassium sensibles à mitochondrial joue un rôle important pour empêcher la mort des cellules nécrotiques et l’apoptsis pendant l’ischamie / des phénomènes de reperfusion en raison de prévenir les pores de transition de perméabilité mitochondriale (MPTP) ouverture, surcharge de calcium intracellulaire et perte d’espace intermembranaire mitochondrial.
Il existe des preuves cliniques d’effets bénéfiques d’un groupe de boissons appelées ouvre-canaux de potassium que la colud soit un nouvel outil thérapeutique contre la maladie d’ischancs cardiaques et Sa visession.
Mots-clés: Confonditionnement ischémique Canaux de potassium sarcolémique et mitochondrial, surcharge de calcium, stress oxydatif, apoptose, pore de transition de perméabilité mitochondriale.
Introduction
à ce jour , le traitement de la maladie cardiaque de l’origine ischémique s’est concentré sur la prévention des dommages ischémiques, augmenter la contribution de l’oxygène à la zone du myocarde en danger ou Diminution de la consommation d’oxygène de myocytes cardiaques.
De même, il est également connu que l’exposition des myocytes à de brèves événements ischémiques à plusieurs reprises, produit une protection contre les événements d’ischémie post-durables, processus appelé pré-adresse ischémique (1 ).
Actuellement, l’intérêt scientifique est concentré dans la recherche des mécanismes cellulaires impliqués dans la cardioprotection contre les événements d’ischémie / de reperfusion, parmi lesquels les canaux de potassium sensibles à l’ATP (K ATP) semblent avoir un rôle central (1- 3).
Dans cet article, les concepts de la physiologie cellulaire et moléculaire liés au rôle de l’ATP K sont examinés dans la défense ischémique du myocarde.
structure et fonction des canaux de potassium cardiaque .
potassium (K +) est la deuxième cation la plus abondante de l’organisme. Un adulte de 70 kg contient environ 4200 MEQ Potassium ou environ 50 MEQ / kg chez les hommes et 40 MEQ / kg chez la femme, en tenant compte de l’ajustement par l’adipocité et la masse corporelle. Le contenu K + de l’organisme décline avec l’âge d’environ 2 mètres / kg pour 10 ans en diminuant la masse musculaire.
environ, 98% de K + se situe dans le liquide intracellulaire, avec une concentration de 150 MEQ / L, tandis qu’en dehors des cellules est de 4 MEQ / L, avec une plage de normalité étroite comprise entre 3,5 et 5,5 MEQ / L.
La quantité K + ingérée dans les pays occidentaux est de près de 50 à 100 MEQ / jour ou plus ou moins de 0,7 à 1,3 mètre / kg de poids corporel par jour. Par exemple, chez une personne qui consomme 80 Meq dans une journée, le rein sera responsable de l’excrétation de 70 Meq, du tractus gastro-intestinal 9 Meq et de la peau d’environ 1 MEQ.
potassium est dans une balance corporelle étroite , dont une partie est due à une redistribution entre les différents compartiments cellulaires, que cedit ion soit traduit du milieu intracellulaire à l’extracellulaire ou inversement (4). Pour qu’un tel phénomène se produise, la cellule utilise des protéines transmembranaires qui fonctionnent comme des canaux.
Les canaux d’ions ont trois propriétés essentielles:
1) un tunnel central ou un pore à travers lequel flux d’ions par rapport à leur gradient électrochimique.
2) Un filtre de sélectivité, qui dicte quel ion sera autorisé à passer à travers le pore, appelé région P
3) une structure qui exerce la fonction de porte en contrôlant la probabilité de conformation de l’ouverture et de la fermeture du canal et, par conséquent, la perméabilité de ladite protéine (4,5,6,7).
Les canaux de potassium sont composés d’alpha et Beta Subunits. Responsable de la conduite des ions à travers la bicouche lipidique est la sous-unité alpha.En fonction de la topologie et le nombre de régions p (sélectivité de pores), un classement des canaux potassiques (4,7,8) a été établie.
Ainsi, par exemple, les canaux potassiques qui augmentent o Diminution leur perméabilité en fonction de la tension transmembranaire, ils ont six segments transmembranaires (TM) et d’un pore (1 p), et sont appelés en fonction de la tension des canaux K + (KV, figure 1, a); Ceux qui présentent deux TM-1P sont des canaux qui permettent le passage de potassium vers intracellulaire lorsque la membrane de plasma est à une tension plus négative que le potentiel de l’ équilibre et sont fermés au potentiel plus positif, si elles sont appelées, les canaux potassiques redresseurs vers l’ intérieur (Kir , K Intérieurement Rectifyer) (7,9,10,11,12,13).
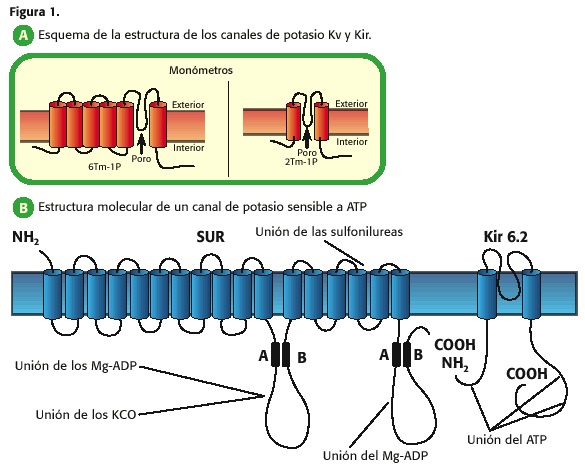
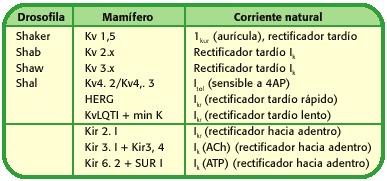
chacune de ces familles de canaux potassiques peuvent à leur tour présentent sous – familles, selon l’électrophysiologie du canal ou ligand qui médie l’ouverture de ces protéines, le tableau 1.
Tableau 1. nomenclature et la structure des canaux potassiques (14).
dans les canaux redresseurs sont à l’ intérieur de deux sous – familles importantes dans la physiologie cardiaque: 1) les canaux K régulés par la protéine G, qui sont activés par le R. Muscarinian Ecetrators M3 (KACH) et 2) les canaux sensibles à l’ ATP (KATP), ces dernières objet central de l’examen (7,9,10,13).
La fonction du k ATP Il est préférable connu dans les cellules bêta-pancréatiques, où change couplant dans l’excitabilité électrique de la membrane plasmatique et la libération d’insuline, par rapport à la concentration en glycémie. Cependant, ils ont également été impliqués dans la protection neuronale dans des événements ischémiques et l’ épilepsie, la régulation de tonus pulmonaire et systémique), le glucose recovenity dans le muscle squelettique et la protection contre l’ ischémie De myocytes cardiaques, le sujet de cet examen (10).
Ces propriétés découlent de la capacité des canaux KATP pour coupler le métabolisme cellulaire à l’activité électrique, la détection des changements dans le cytosol des niveaux d’ ATP et l’ adénosine diphosphate (MG-ADP), fonctionnant l’ATP en tant que bloqueur des canaux, et le MG- ADP comme un activateur ou promoteur de son ouverture (10,11).
le pore des canaux de potassium est un tétramère et parce que le KIR sont 2 TM-1P, quatre de ces unités étant réunis pour former le pore, libellé au niveau cardiaque KIR 6.2 et KIR 6.1. En outre, il existe une autre protéine appelée récepteur de sulfonylurée (sort, récepteur de sulfonylurée, figure 1, B (15))
qui régule l’ouverture ou la fermeture du pore KIR 6.2 ou KIR 6.1: L’ATP inhibe le canal déjà qu’il se joint à la sous – unité KIR 6, tandis que le MG-ADP il active par son interaction avec la sous – unité du Sud (10,11,12,13,14).
fonction centrale de la mitochondrie dans La cardioprotection
mitochondria se trouve dans presque toutes les cellules à l’exception de Hématis et leur nombre varie en fonction du type de cellule; Par exemple, chaque hépatocyte comporte 1 022 à 2 000 mitochondries, qui mesurent 3 μm de long environ.
Mitochondria possèdent deux membranes, un interne externe et d’autres internes, ce qui entraîne des compartiments intermembranaires et la matrice mitochondriale. Dans la matrice mitochondriale et dans la membrane interne, il s’agit là où la plupart des activités liées à la chaîne respiratoire (16) sont développées. Dans la mitochondria, plusieurs protéines jouent des fonctions particulières, que la promotion de la cardioprotection ou du détriment de la fonction cardiaque; et ils sont reliés entre eux par le KATP par des mécanismes différents
Dans les plus importants sont:.
1) les pores de la perméabilité de transition (MPTP, la perméabilité mitochondriale pores de transition) qui est constitué par:
– canal d’anions dépendant de la tension (VDAC, Voltage – Dependent anion Channel), qui est situé dans la membrane mitochondriale externe (MME)
– le transporteur de nucléotides d’ adénine (ANT, adénine nucléotide Translocator), situé dans la membrane interne (MMI)
– la créatine kinase (CK), dans l’espace intermembranaire (EIM)
2) les contre-transporteurs de Na + / H +, Na + / Ca ++, K + / H +, H + / pyruvate, le Ca ++ cotransporteur et les protéines bien connues de la chaîne respiratoire, la bêta-oxydation et le cycle de Krebs (17,18) ( Figure 2).
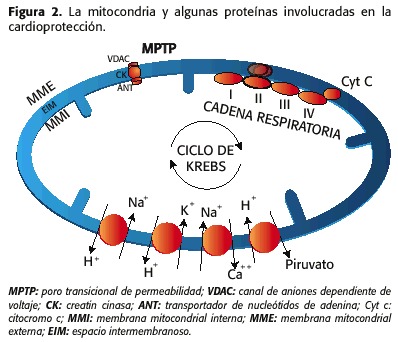
événements plus importants impliqués dans la genèse de l’ ischémie / contrecoup sont:
1. Changements dans le volume de la matrice mitochondriale
2. Le statut EIM
3. La perméabilité du MMI
4. La rupture du MME et
5. La surcharge de calcium-sodium intramitocondrique.
Physiologie mitochondriale de la manipulation de calcium.
Calcium joue un rôle fondamental dans la physiologie mitochondriale, dont les concentrations sont dans des gammes étroites. Le calcium peut être recapté vers la matrice mitochondria à travers la MMI par trois mécanismes:
1) une pompe de calcium, appelée uni-calcium ou up,
2) un canal qui permet L’entrée rapide du calcium, connue sous le nom de protéine de transport rapide du calcium ou de la RAM (absenquez l’absorption de Ca ++) et
3) une rivière ou Ryr (récepteur de la ryanodine).
sur l’autre Main, la sortie de calcium de la matrice mitochondriale est due à:
1) le contre-convoyeur NA + / CA ++, dans des conditions physiologiques
2) le MPTP, dans des conditions pathologiques (figure 3)
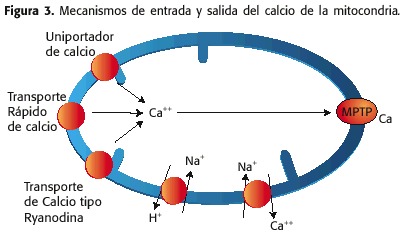
fonction primaire du calcium au niveau de La mitochondria est la stimulation de la chaîne respiratoire à plusieurs niveaux, entraînant une augmentation de la production d’ATP et de son exportation au cytosol pour répondre à la demande métabolique de MyOcit.
sait également que le processus de phosphorylation oxydatif génère une PE Quantité de radicaux libres en tant que produit de complexes I, Coenzyme Q et Complexe III, et que loin d’être nocifs, plutôt nécessaires à la transduction du signal de plusieurs voies métaboliques intramitocondoïques (19).
Mécanismes qui favorisent Surcharge de calcium intramitocondrique. Au cours de l’ischémie du myocarde, l’augmentation de la glycolyse provoque une accumulation progressive de l’acide lactique, une diminution de pH mitochondrial et qui exercera une rétroaction négative que finalement
inhibe la glycolyse et la production d’ATP. L’activation de l’anti-go + / h + anticipateur qui extrude les hydrogénions de la mitochondria recherche la normalisation du pH intramatricial, mais dans le processus une surcharge de sodium sera encouragée, qui ne peut être pompée de la cellule, car Le NA + / K + -ATPASE est inhibé par la perte d’ATP (19). Par conséquent, l’activité du contre-convoyeur Na + / CA ++, qui exporte généralement du calcium de la mitochondria et de la cellule, est diminuée ou même agissant inversement (car il s’agit d’un mécanisme de transport actif secondaire) de sorte que dans les différents compartiments cellulaires initie un retour positif qui favorise grandement une surcharge de calcium. À ce stase, l’homéostasie ionique ne peut pas être maintenue, les concentrations intracellulaires de sodium et de calcium augmentent progressivement, avec une diminution des nucléotides d’adénine et une augmentation du phosphate.
En outre, l’augmentation de calcium mitochondrial favorise la formation radicale libre dans la mitochondrime Par les mécanismes suivants:
1) La surcharge de calcium mitochondrial favorise une augmentation du flux d’électrons dans la chaîne respiratoire
2) stimule également l’oxyde nitirique synthétase, formant de l’oxyde nitrique (NO)
3) est connu qu’elle n’inhibe pas la chaîne respiratoire au niveau complexe IV Que
4) augmente la formation de radicaux libres (ROS, espèces d’oxygène réactives) par cycle q
5) Les complexes I et II de la chaîne respiratoire peuvent également être inhibées par l’augmentation du calcium et non, contribuant encore plus à la formation de ROS
6) dissocier également le cytochrome c de protéines cardiolipin dans MMI et enfin Mo
7) L’augmentation du calcium favorise l’ouverture du MPTP, permettant la libération du cytochrome, par le biais de MME (19).
malgré tout ce qui précède, pendant l’ischémadiale Le pH (IMM) reste faible et ce phénomène, comme on le discutera dans les sections suivantes, est suffisamment puissant pour maintenir le MPTP fermé, tant que l’événement d’ischemia ne prolonge pas ou ne surmonte pas la réperfusion redoutée.
Le pore de perméabilité transitoire (MPTP)
Le MPTP a été décrit par Haworth et Hunter il y a plus de 25 ans, cependant, il y a quelques années qu’il y a quelques années que sa fonction avait une grande importance dans les états d’ischémie reperfusion (20,21,22). Dans un état d’équilibre ou une homéostasie mitochondriale, le MPTP est fermé, la distance
entre MME et MMI est optimale et il y a ainsi un couplage entre le VDAC-CK-ant, ce qui permet que le mécanisme énergétique principal de la Mitochondria est le système de créatiche-phosphocréatine et d’autre part de celui de ATP / ADP (Figure 4) (23). De plus, la perméabilité de l’ANT-VDAC est faible dans cet état et les canaux de potassium dépendant de l’ATP sont fermés (18,23,24).
d’autre part, l’antipersporter k + / h + prend du potassium de la matrice en échange d’une hydrogénión, et a également un faible cinétique dans des conditions fixes, afin de garantir le maintien de la phosphorylation oxydative, un volume de matrice adéquat et un potentiel de membrane mitochondriale négatif, car comme on le sait, le potentiel d’hydrogénions entre l’EIM et le La matrice mitochondriale est maintenue à l’imperméabilité du MMI aux différents ions, ce qui permet à la génération d’ATP par le complexe V du V dans le MMI.
En conclusion, d’un point de vue physiologique, de la réglementation du volume de la matrice mitochondriale a des conséquences importantes sur le métabolisme de l’énergie myocus et l’état étanche du pore contribue à ladite homéostasie (17,24,25).
Le pore de perméabilité transitoire pendant l’ischémie et lors de la répercussion . Dans des conditions d’ischémie myocardique, le MPTP s’ouvre sur une quantité inférieure, permettant au MMI d’être perméable à une molécule < 1,5 kDa. Deux conséquences importantes se produisent alors. Tout d’abord, les protéines intramitocondiales ne peuvent pas passer par le pore et exercer une pression colloïdo-meuble dans le gonflement matriciel. Le MMI en raison de sa consistance ne souffre pas de lyse, cependant, un tel phénomène se produit dans MME, avec la libération de protéines à l’EIM, telle que le cytochrome C et le facteur inducteur de l’apoptose qui joue un rôle crucial dans la mort cellulaire par ce mécanisme . Deuxièmement, le MMI devient perméable aux protons, qui ont inculpéré la chaîne respiratoire, diminuaient la production d’ATP et troisième, le fonctionnement inverse de l’ATP Synthetase est favorisé, c’est-à-dire au lieu de synthétiser l’ATP, a favorisé l’hydrolyse de ce souci de maintenance Un gradient de H + et un potentiel membranaire mitochondrial négatif (18 26,27,28,29).
Cependant, dans une telle situation, les concentrations d’ATP déclinent rapidement, conduisant à une altération ionique de l’homéostasie métabolique et l’activation des enzymes qui favorisent la dégradation, telles que les phospholipases, les nucléases et les protéases. Sauf si la fermeture de la MPTP n’arrive, ces changements provoquent des dommages cellulaires irréversibles, ce qui entraîne une nécrose cellulaire.
Un facteur clé dans l’ouverture du MPTP est la surcharge de calcium intralatricielle, en particulier lorsqu’elle est accompagnée de stress oxydant, d’une épuisement de Nucléotides adénosines, fortes concentrations de phosphate ionique et dépolarisation du MMI, microenvironnement présenté dans l’ischémie. Par conséquent, l’ouverture du MPTP est une étape critique de la transition de dommages de cellules réversibles à irréversibles (18,23).
Comme mentionné ci-dessus, le MPTP est formé par l’ANT-VDAC et de l’autre La protéine, appelée cyclophiline-d (CYP-D), qui doit rejoindre la fourmi pour provoquer son ouverture, l’union qui est favorisée par l’augmentation de la concentration en calcium, par l’épuisement des réserves de nucléotidiques de l’adénosine, en augmentant les IMM et les altérations de volume matriciel. Cependant, un puissant inhibiteur d’ouverture de pores est la réduction du PHMI, un phénomène qui survient pendant l’ischémie, établissant un équilibre entre les facteurs favorisant son ouverture (surcharge de calcium mitochondrial, épuisement de l’ATP, élévation de stress oxydatif) et ceux qui le gardent fermés ( sous PHMI), figure 5.
donc tous les deux, pendant l’ischémie le MPTP Ouvre mais à faible quantité, car la micro-nation cellulaire n’est pas optimale pour qu’un tel phénomène se produise. D’autre part, lors de la reperfusion, il existe une série de phénomènes intramitocondriaux qui favorisent l’ouverture du MPTP encore plus que pendant l’ischémie. Lorsque la reperfusion se produit, la mitochondria est à nouveau capable de respirer et de générer un potentiel membranaire qui permet la synthèse de l’ATP, avec l’augmentation de la production de radicaux libres tels que des produits de la chaîne respiratoire, le PHMI commence à augmenter et toujours persister le Surcharge de calcium et la déviation de l’ATP. Par conséquent, à ce stade, il est favorisé l’ouverture du MPTP (30,31,32,33,34,35) (Figure 6). En fonction de la proportion de pores ouverts et du temps restant dans un tel état, une nécrose ou une apoptose (18,23,24) ira à la promotion (figure 7).
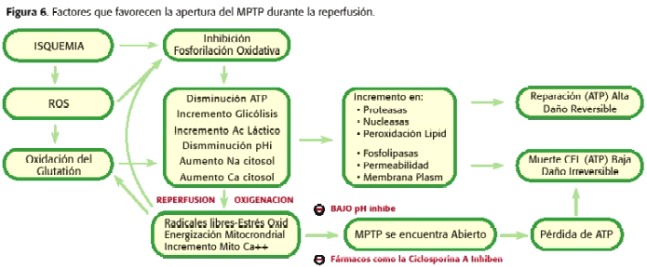
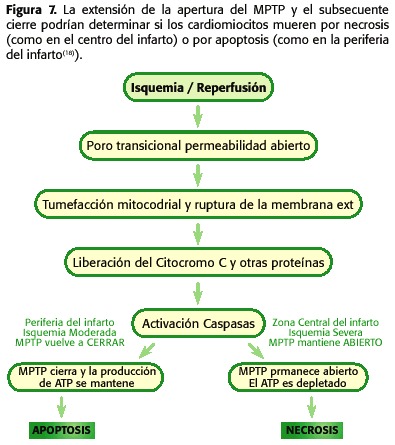
De cette manière, la mitochondria devient un objectif thérapeutique, soit la promotion de l’inhibition directe du MPTP, soit indirectement, en empêchant les phénomènes qui favorisent son ouverture, telle que
1) la surcharge de calcium pouvant être obtenue avec la propofol36 ou avec l’augmentation des concentrations de Mg ++ qui inhibe le contre-convoyeur Na + / Ca ++ et les canaux de calcium Type L (37)
2) Améliorer la bioénergétie mitochondriale ou le volume de matrice, les deux phénomènes propitichés à des médicaments ouverts de canaux de potassium (kco, ouvre-canaux de potassium) voir ci-dessous
3) Maintenir un pH faible pendant la reperfusion, par exemple, en inhibant l’antipersportador Na + / H + avec des médicaments tels que l’amilorure ou en ajoutant du pyruvate (38) à la mitochondrie qui encourage l’augmentation de l’acide lactique.
tout à Nterta ouvre une immense porte dans la recherche en sciences fondamentales et en thérapeutique pour les patients souffrant d’ischémie myocardique ou de conséquences.
canaux de potassium sensibles à l’ATP: sa fonction dans la prévention de l’ouverture de la MPTP Mitochondrial et Dans la membrane sarcoplasmique
Pendant les situations de stress myocardial, dans laquelle l’ATP est réft, les canaux de K + augmentent leur statut d’ouverture avec des conséquences bénéfiques pour le microenvironnement de MyOcit (39,40, 41).
L’ouverture du Kitchondrial K ATP provoque une augmentation modeste de l’influence K + à l’intérieur de la matrice, ce qui provoque deux effets différents en fonction de l’état bioénergétique de cardiomyocyte. Premièrement, lorsque la cellule est au repos, le potentiel membranaire mitochondrial est élevé et l’entrée d’ions potassium favorise la tortue mitochondriale et l’alcalisation de la matrice, ce qui encourage une légère augmentation de la production de radicaux libres. Deuxièmement, si la cellule est dans l’état d’ischémie, la mitochondria est déployée (faible potentiel), qui favorise la sortie de K +, un phénomène qui est neutralisé par les canaux de K ATP et donc la contraction du volume de la motrice qui serait Sinon, on se produit (42).
Le revenu de potassium à la matrice mitochondriale génère une légère production de radicaux libres (ROS) et, comme il est démontré, ils jouent un rôle important en tant que secondes messagers dans une variété de signaux intracellulaires (39). Cette production de la cellule de repos favorise également la récupération de K + en échange par un H + (Antitransporter K + / H +), qui crée le gradient pour l’échange d’un phosphate (PI) par le cotransporter électro-utilitaire PI / H + . La récupération PI est beaucoup moins que celle de K +, car le PI est présent dans une concentration beaucoup moins que la K +. Pour cette raison, le pH de la matrice est toujours augmenté lorsque le volume de la matrice augmente également, les phénomènes promus par la récupération PI et K + (23).
lorsque les canaux n’ont pas encore entré dans la fonction de KATP lors d’événements ischémiques, la matrice mitochondriale subit une contraction et une augmentation de l’EIM, avec la désunité du VDAC de la fourmi par le CK, qui augmente la conductance à l’échangeur nucléotidique par le VDAC et la fourmi, contrairement à ce qui est se passe dans le repos, contribuant ainsi à la déflexion de la matrice et de la cytosoline ATP (Figure 3). Par conséquent, l’ouverture de la KATP pendant l’ischémie, par exemple promue par le diazoxyde, maintient le volume de l’EIM, réduit le taux de perte d’ATP, réduit le taux de dégradation des nucléotides d’adénine de manière à ce qu’ils existent des réserves ADP pour la suite La phosphorylation lors de la reperfusion et finalement, réduit les modifications apportées au potentiel de la membrane mitochondriale et à l’accumulation de CA ++, empêchant la surcharge de calcium, car l’ATP reste dans des concentrations appropriées pour un fonctionnement minimal de Na + / K + ATPASA et d’autres pompes.
Ces effets préservent la fonction mitochondriale et peuvent ainsi être confrontés à la reperfusion avec de meilleurs résultats physiopathologiques. Pendant la reperfusion, l’ouverture ultérieure des canaux KATP permet à la compartimentation des nucléotides d’adénine, ce qui signifie que la première source d’énergie est phosphate de la phosphocréatine et non l’ATP (faible conductance du VDAC (23), la figure 3).
Par conséquent, l’activation des canaux ATP mitochondriaux pendant l’ischémie-reperfusion empêche la formation du pore de perméabilité transitoire en inhibant la contrainte oxydative, déclenchée par le revenu de la CA ++ (18,23,24).
Vanden Hoek et al. Ils ont montré que l’augmentation des radicaux libres dans les mitochondries lors de la condition préalable ischémique était nécessaire pour protéger le myocus contre une contrainte oxydante ultérieure lors de la reperfusion.C’est ainsi que cela a été hypothéqué qu’il existe deux étapes dans la libération de radicaux libres, la première qui contribue à l’ouverture des canaux de potassium sensibles à l’ATP et à une suite lors de la répercussion, qui est nocif et déclencheurs rénové et apoptose cardiaque ( 42). Dans la membrane sarcolémique du myocyte cardiaque et du muscle lisse, l’ouverture de canaux de potassium contient également des effets protecteurs.
Hyperpolarisation des cellules qui empêche la surcharge de Ca ++ intracellulaire, diminuant la durée du potentiel d’action, limitant la cellule cellulaire Dommages causés et préservant les réserves d’énergie cellulaire et donc la survie de MyOcit. De plus, cette séquence peut être à la fois aiguë et chronique (8,42,44). Au niveau vasculaire, l’ouverture de ces canaux favorise la vasodilatation, en raison de l’hyperpolarisation cellulaire et de la réduction de l’entrée de CA ++, avec un flux coronaire accru et une diminution du post-charge.
perspectives cliniques
En raison des propriétés combinées de cardioprotection et de vasodilation, des médicaments permettant l’ouverture de canaux de potassium (KCO), pourraient être considérés pour certaines conditions cardiaques (tableau 2). Celles-ci incluent la protection du myocarde sous la circulation extracorporelle, la préservation du cœur des donateurs dans la transplantation cardiaque, le traitement de la maladie ischémique cardiaque, de l’hypertension artérielle systémique et pulmonaire, des maladies vasculaires périphériques et des arythmies liées à une repolarisation anormale (41-50).
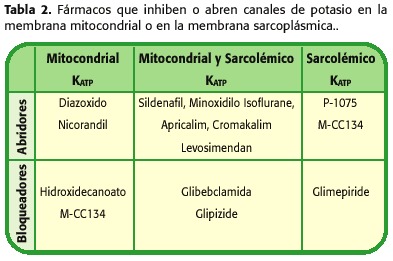
dans la chirurgie cardiaque, le KCO pourrait jouer un rôle important à jouer Solutions Cardioplegia. Dans plusieurs modèles de chirurgie cardiaque avec une circulation extracorporelle, plusieurs KOC, y compris Nicorandil, Aprikalim et Pinacidil, ont favorisé une plus grande cardioprotection qu’avec la cardioplexie conventionnelle (51,52).
chez les patients ayant effectué des ponts chirurgir (contournement) coronary, Le temps nécessaire pour atteindre l’arrestation cardiaque, les changements dans le segment ST après le serrage aortique, les niveaux de plasma du CPK-MB et la dose d’agents inotropes étaient tous mineurs que le groupe traité avec Nicorandil par rapport aux commandes traitées avec une thérapie conventionnelle ( 53).
L’utilisation de solutions cardioplexiques avec kco tels que le diazoxyde, le minoxidyle (54), la propofol (qui inhibe la surcharge de calcium ++ (36)), le magnésium (qui inhibe le surcharge de calcium37), les numérosiens (qui favoriser la cardioprotection par des voies de signalisation cellulaire qui convergent avec celles de KCO (23.55)), pyruvate (qui encourage l’acidose intracellulaire et Normalisation du grade de pH par la fermeture MPTP (38)), les inhibiteurs de contre-convoyeur Na + / H ++ tels que l’amylorure (qui évitent également la perte de l’effet de protection bas de la faible), adénosine (qui est également un kco , agissant à travers des protéines G, empêche la surcharge de calcium, car la cardioplegie hyperpothémique favorise (52,56) objectifs pour la préservation de la fonction cardiaque dans les chirurgies à haut risque et peut éviter les complications potentiellement mortels tels que le syndrome de dépenses post-pompes et de nombreux d’autres, avec la réduction des coûts économiques en réduisant les séjours à l’hôpital.
D’autre part, l’utilisation d’antioxydants ne semble être bénéfique que dans certaines circonstances, déjà que son utilisation dans des essais de laboratoire a bloqué la pré-revêtement ischémique , parce que les radicaux libres sont libérés en quantité inférieure (23,25).
dans le syndrome coronarien aigu, la valeur Du KCO est mieux documentée cliniquement avec Nicorandil qui s’est avéré être avantageux avec des effets indésirables minimes sur la manipulation d’une angine stable et instable (57). Dans une étude multicentrique impliquant plus de 5 000 patients atteints de stables d’Angor, l’utilisation à long terme de Nicorandil était associée à la réduction des événements cardiovasculaires tels que la mort cardiaque, l’infarctus du myocarde et l’hospitalisation en raison de la douleur thoracique (58,59).
Les patients atteints d’une colère instable, le Nicorandil a ajouté à un traitement anti-ischémique agressif réduit des épisodes d’ischémie / nécrose myocardique et d’arythmie par rapport aux patients traités avec une thérapie conventionnelle. Chez les patients subissant une angioplastie coronaire, le Nicorandil a prédominé le cœur, une hémodynamique coronaire améliorée et une viabilité du myocarde préservée avant la reperfusion (60,61,62).
Nicorandil diminue également la précharge et la postelette, elle augmente la libération de nitrique. L’oxyde de cellules endothéliales et, contrairement à la nitroglycérine, ne développe pas la tolérance à ses effets antivols (63).
Dans l’angine Vasospastal, le Nicorandil est un vasodilatateur puissant et a été démontré pour réduire les épisodes de la variante angine, diminue le segment ST et améliore la perfusion coronaire. Le KOC a également démontré de meilleurs résultats dans la chirurgie de pontage coronaire utilisant des greffes artérielles (artères thoraciques internes, gastropiépploïques, radiales), car elle évite le vasospasme ultérieur (64,65,66)). L’expérience et les données scientifiques sont nécessaires avec l’utilisation de KCO dans ces situations cliniques.
L’activation de la KATP sarcolémique est responsable du courant électrique soulignant l’élévation de segment ST, l’indicateur électrocardiographique classique des dommages ischémiques de transuroménums Miocardial ( 41). Les patients atteints de diabète de diabète traité avec des sulfonylurées et qui sont infarctus aiguë au myocarde, présentent une magnitude atténuée à St altitude, ce qui rend difficile le diagnostic initial, car les sulfonylurées sont des inhibiteurs des canaux KATP à charge (41).
CONCLUSION
Comme pratiquement toutes les maladies humaines, la thérapeutique de la maladie ischémique cardiaque repose sur de nouvelles connaissances en physiologie cellulaire et physiopathologie. La modulation de canaux KATP est un processus critique dans l’homéostasie métabolique de la cellule et, selon la recherche biomédicale dilue de nouveaux concepts sur la structure, la fonction, la régulation et la sélectivité des tissus de ces canaux, de nouveaux agents thérapeutiques peuvent être développés avec d’énormes avantages pour la population à risque.
références
1. Ardéhali h, O’Rourke. Canaux KATP mitochondriaux dans la survie des cellules et la mort. J Cellule MOL CARDIOL 2005; 39: 7-16.
2. Klonner Ra, Bolli R, Marban E, ReiniliB L, Braunwald E. Implications médicales et cellulaires d’étourdissements, d’hibernation et de préconditionnement: un atelier de la NHLBI. Circulation 1998; 97: 18: 48-67.
3. Murray CE, Jennings RB, Reiner Ka. Précondictionnant avec ischémie: retard de la lésion cellulaire mortelle dans le myocarde ischémique. Circulation 1986; 74: 11: 24-36.
4. Cogan mg. Liquides et électrolytes: physiologie et physiopathologie. Manuel moderne. Mexique, 1993; 145-168.
5. Sansom MSP, Shrivastava IH e à. Canaux de potassium: structures, modèles, simulations. Biochimica et Biophysica Acta 2002, 1565: 294-307.
6. Biggin PC, Roosild T, Choe S. Structure du canal de potassium: domaine par domaine. Opinion actuelle Biologie structurelle 2000, 10: 456-461.
7. Giblin JP, Leaney JL, et Tinker A. L’assemblage moléculaire des canaux de potassium sensibles à l’ATP: déterminants sur la sous-unité formant. J Biol Chem 1999, 274: 22652-22659.
8. Structures de canal de potassium Choe S.. Nat Rev Neuroscience 2002, 3: 115-121.
9. Loussouarn G, Rose T, Nichols Cg. Base structurelle de la récupération de canal de potassium rectifiant vers l’intérieur. Tendances Cardiovasc Med 2002; 12: 253-258
10. Nishida M, Mackinnon R. Base structurelle de rectification intérieure: pore cytoplasmique du redresseur intégré à la protéine G Girk1 à 1,8 à la résolution. Cellule 2002; 111: 957-965.
11. Yamada M, Inanobe A, Kurachi Y. G Régulation de la protéine des canaux d’ions de potassium. Commentaires pharmacologiques 1998; 50: 724-747.
12. Bichet D, Haass F, Yeh Jan L. Fusionner des études fonctionnelles avec des structures de redresseur intérieur K +. Nature Avis 2003; 4: 957-67.
13. Aguilar-Bryan L, Clement IV J, González G, Kunjilwar K, Babenko A, JJ Bryan vers la compréhension de l’assemblage et de la structure des canaux KATP. Commentaires physiologiques 1998; 78: 227-242.
14. Elizari M, Chie P. Arythmies cardiaques: fondamentaux cellulaires et moléculaires, diagnostic et traitement. Panamericana, Buenos Aires, 2003; 31-40
15. Campbell JD, Sanson MSP, Règlement sur le canal de potassium Ashcroft F.. Nature Avis 2003; 11: 1038-1042.
16. Méndez Je, Zeledón SF, Zamora JF, Cortés VA. Une approche de la cinétique d’oxygène partie 1. Rev Costar-Costardiol 2004; 6: 27-32.
17. Suleiman M, Haletrap AP, Griffiths E.J. Mitochondria: une cible de protection du myocarde. Pharmacologie et thérapeutique 2001; 89: 29-46.
18. Haletrap AP, Clarke J, Sabzali A, Javadov A. Présentation de perméabilité mitochondriale Ouverture de la perméabilité lors de la répercationalisation du myocarde: une cible de cardioprotection. Cardiovasc 2004; 61: 372-385.
19. Brookes P, Yoon et, Robotham J et al. Calcium, ATP et ROS: Triangle d’amour mitochondrial. Suis J Physiol Cell Physiol 2004; 287: C817-C833.
20. Hunter Dr et Haworth Ra. La transition membranaire induite par la CA ++ dans la mitochondria I. Les mécanismes de protection. Arch Biochem Biophys 1979; 195: 453-459.
21. Hunter Dr et Haworth Ra. La transition membranaire induite par la CA ++ dans la mitochondria II. Nature du site de déclenchement CA ++. Arch Biochem Biophys 1979; 195: 460-67.
22. Hunter Dr et Haworth Ra. La transition membranaire induite par le CA ++ dans la mitochondria III.Relase Ca ++ transitionnelle. Arch Biochem Biophys 1979; 195: 468- 77.
23. Garlid KD, Dos Santos P, Xie ZJ, Costa A, Paucek P. Mitochondrial Potassium Transport: Le rôle du canal K + sensible à la CTP mitochondrial dans la fonction cardiaque et la cardioprotection. Biochem Biophys Acta 2003; 1606: 1-21.
24. MacFalleras E, Liem d et al. Fonction mitochondriale: le cœur de la préservation du myocarde. J Lab Clin Clin Med 2003; 142: 141-9.
25. Facundo H, Fornazari M, Kowaltowski A. Protection des tissus médiée par des canaux K + mitochondrial. Biochem Biophys Acta 2005; 1701: 1-11.
26. Haltrap AP. La transition de perméabilité mitochondriale: son mécanisme moléculaire et son rôle dans la lutte contre la réperfaction. Dans: Brown GC, Nicholls DG, Cooper CE, Éditeurs. Mitochondria et mort cellulaire. Symposium de la société biochimique. Londres: Press Press; 1999, vol. 66, p. 181- 203.
27. Halestrap AP, McStay GP, Clarke SJ. Le complexe de pores de la transition de perméabilité: une autre vue. Biochimie 2002; 84: 153-66.
28. Halestrap AP, Kerr PM, Javadov S, Woodfield Ky. Élucidant le mécanisme moléculaire du pore de la transition de perméabilité et son rôle dans la lutte contre la réperfaction du cœur. Biochim Biophys Acta 1998; 1366: 79-94.
29. Halestrap AP, Brenner C. La translocase de nucléotide adénine: une composante centrale du pore de transition de perméabilité mitochondriale et du joueur clé dans la mort cellulaire. Cour Med Chem 2003; 10: 1507-25.
30. Duchen MR, McGuinness O, Brown La, Crompon M. Crompton M. sur l’implication d’une cyclosporine – un pore mitochondrial sensible dans la lutte contre la réperfaction du myocarde. Cardiovasc res 1993; 27: 1790-4.
31. Lemasters JJ, Nieminen Al, Qian T, Trost LC, Herman B. La transition de perméabilité mitochondriale dans une blessure toxique, hypoxique et de réperfaction. Mol Cell Biochem 1997; 174: 159-65.
32. Lemasters JJ, Trollinger Dr, Qian T, Cascio Nous, Ohata H. Imagerie confocale de CA2 +, pH, potentiel électrique et perméabilité à la membrane dans des cellules vivantes. Méthodes Enzymol 1999; 302: 341-58.
33. Xu MF, Wang YG, Hirai K, Ayub A, Ashraf A. Le préconditionnement du calcium inhibe la transition de perméabilité mitochondriale et l’apoptose. Suis J J Physiol 2001; 280: H899- 908.
34. Miyata H, Lakatta EG, Stern MD, Silverman HS. Relation du calcium gratuit mitochondrial et cytosolique à la récupération de myocytes cardiaques après une exposition à l’anoxie. CRC RES 1992; 71: 605-13.
35. Griffiths EJ, Ocampo CJ, Savage JS, et al. Pistes de transport de calcium mitochondrial lors de l’hypoxie et de la réoxygénation dans des cardiomyocites simples de rat. Cardiovasc res 1998; 39: 423-33.
36. LIM KHH, Modi P, Nicholson E, et al. Un modèle de cochon d’arrestation cardioplegique au sang chaud pour enquêter sur les effets cardioprotecteurs de la propofol. J Physiol 2001; 536P: 82p.
37. Headrick JP, McKirdy JC, Willis RJ. Effets fonctionnels et métaboliques du magnésium extracellulaire dans le myocarde normoxique et ischémique. Am J Physiol 1998; 275: H917-29.
38. Maillet rt. Pyruvate: protecteur métabolique de performances cardiaques. Proc Soc Exp Biol Med 2000; 223: 136- 48.
39. Oldenburg O, Cohen M et al. Canaux KATP mitochondriaux: rôle dans la cardioprotection. CARDIOVASC RES 55: 429-437, 2002.
40. Minners J, McLeod C, SACK N. Plasticité mitochondriale dans le préconditionnement ischémique classique Au-delà du canal KATP mitochondrial. Cardiovas Res 2003; 59: 1-6.
41. Kane G, Liu X, Yamada S, Olson T, Terzic A. Canaux KATP cardiaques de la santé et des maladies. J Cellule MOL CARDIOL 2005; 38: 937-943.
42. Wang Y, Haider H, Ahmad N, Aschraf M. Mécanismes par Wich Katp Canal Openers produisent une cardioprotection aiguë et retardée. Pharmacologie vasculaire 2005; 42: 253-264.
43. Vanden Hoeck T, Becker L.B, Shao Z, Schumker P. Les espèces d’oxygène réactives libérées de mitochondries lors d’une brève hypoxie induisent le préconditionnement dans des cardiomyocytes. J Biol. Chem. 1998; 273: 18092-98
44. Jahangir A, Terzic A. Katp Channel Therapeutics au chevet. J Cellule MOL CARDIOL 2005; 39: 99-112.
45. Kane GC, Behfar A, Yamada S, Perez-Terzic C, O’Cochlain F, et al. Hoenicke em, Sun XW, étrange rg, Damiano rj. Préservation du cœur des donateurs avec une nouvelle solution hyperpolarisatrice: protection supérieure par rapport à la solution de l’Université de Wisconsin. J Thorac Cardiovasc Surg 2000; 120: 746-54.
46. Quast U, Guillon JM, Cavantage I. Pharmacologie cellulaire des ouvre-canaux de potassium dans un muscle lisse vasculaire. Cardiovasc res 1994; 28: 805-10.
47. Okada Y, Yanagisawa T, Taira N. BRL 38227 (Levcromakalim) L’hyperpolarisation réduite réduit la sensibilité à Ca2 + d’éléments contractiles de l’artère coronaire canine. Naunynyn Schmiedebergs Arch Pharmacol 1993; 347: 438-44.
48. Donnelly R, Elliott HL, Meredith Pa, Reid Jl. Études cliniques avec l’activateur de canal de potassium Cromakalim dans des sujets normotensens et hypertendus. J Cardiovas Pharmacol 1990; 16: 790-5.
49. Simpson D, Wellington K.Nicorandil: Examen de son utilisation dans la gestion de l’angine stable Pectoris, y compris des patients à haut risque. Drogues 2004; 64: 1941-55.
50. McCully JD, Levitsky S. Mitochondrial ATP-Sensible aux canaux de potassium dans la cardioprotection chirurgicale. Arch Biochem Biophys 2003; 420: 237-45.
51. Kevelaittis e, Oubenaissa A, peynet J, Mouas C, Menasche P. Préconditionner par des ouvre-canaux de potassium sensibles au Mitochondrial – une approche efficace pour améliorer la préservation des greffes cardiaques. Circulation 1999; 100: 345- 50.
52. Steensrud T, Nordhaug D, Husnes KV, Aghajani E, Sorlie DG. Remplacement du potassium avec Nicorandil dans la cardioplegie de l’hôpital de Saint-Thomas froid améliore la préservation de l’énergie et de la fonction dans les coeurs de porc. Ann Thorac Surg 2004; 77: 1391-7.
53. Hayashi Y, Sawa Y, Ohtake S, Nishimura M, Ichikawa H, Matsuda H Contrôlée Administration de Nicorandil Formyocardial Protection du bypass de l’artère coronaire Sous la pontage cardiopulmonaire. J Cardiovas Pharmacol 2001; 38: 21-8.
54. Garlid PD, Pauduit P et al. Effet cardioprotectif du diazoxyde et de son interaction avec des canaux K + sensibles à l’ATP mitochondrial: mécanisme possible ou cardioprotection, CIRC RES 1997; 81: 1072-182.
55. L’interaction Xic Z. Ouabain avec CARDIAC NA / K- ATPASA révèle que l’enzyme peut agir en tant que pompe et sous forme de transducteur de signal. Cellule mol biol 2001; 47: 383-390.
56. Jovanovic A, López J, Alekseev A et al. L’adénosine empêche le chargement de CA21 induit par K1: aperçu de la cardioprotection pendant la cardioplegia. Ann Thorac Surg 1998; 65: 586-91.
57. Simpson D, Wellington K. Nicorandil: un examen de son utilisation dans la gestion de l’angine stable Pectoris, y compris des patients à haut risque. Drogues 2004; 64: 1941-55.
58. Markham A, Plosker Gl, Goa Kl. Nicorandil – une évaluation mise à jour de son utilisation dans la maladie cardiaque ischémique mettant l’accent sur ses effets cardioprotecteurs. Drogues 2000; 60: 955-74.
59. Le groupe d’étude IONA. Essai pour montrer l’impact du Nicorandil en Angina (Iona): conception, méthodologie et gestion. Coeur 2001; 85: E9.
60. Ito H, Taniyama Y, Iwakura K, Nishikawa N, Masuyama T, et al. Nicorandil intraveineux peut préserver l’intégrité microvasculaire et la viabilité du myocarde chez les patients présentant un infarctus myocardial de mur antérieur répercuté. J amick cardiol 1999; 33: 654-60.
61. Schlepper M, Thormann J, Berwing K, Strasser R, Mitrovicv. Effets du Nicorandil sur la perfusion régionale et la fonction ventriculaire gauche. Les médicaments cardiovasc vont 1995; 9: 203-11.
62. Sakatay, Kodama K, Komamura K, Limyj, Ishikura F, et al. Effet salutaire de l’administration de Nicorandil intracoronnaire adjuctifiée sur la restauration du flux sanguin du myocarde et de l’amélioration fonctionnelle des patients atteints d’infarctus aigu du myocarde.M Coeur J 1997; 133: 616-21.
63. Simpson D, Wellington K. Nicorandil: un examen de son utilisation dans la gestion de l’angine stable Pectoris, y compris des patients à haut risque. Drogues 2004; 64: 1941-55.
64. Kaski JC. Gestion de l’angine vasospastique-rôle du Nicorandil. Les médicaments cardiovasc vont 1995; 9: 221-7.
65. Chen JW, Lee WL, HSU NW, Lin SJ, Ting CT, Wang SP, et al. Effets du traitement à court terme du Nicorandil sur l’ischémie myocardique induite par l’exercice et une activité autonome cardiaque anormale en angine microvasculaire. Am j cardiol 1997; 80: 32-8.
66. Akar F, Uydes-Dogan Bs, Tufan H, Aslamaci S, Koksoy C, Kanzik I. La comparaison de la réactivité des artères internes intérieures et gastroépploïques isolées humaines à Levcromakalim: une approche alternative de la gestion du spasme greffé. BR J Clin Pharmacol 1997; 44: 49-56.
Unpartamento de Fisiología, Universidad de Ciencias Médicas (UCIMED), Sabana Oeste, San José, Costa Rica. Teléfono (506) 296-3944, E-mail: [email protected], [email protected]
B Cátetra de Cirugía, hôpital México, UciMed, San José, Costa Rica.
C Cátedra de Cirugía, hôpital México, Université de Costa Rica (UCR), San José, Costa Rica.
D Cátetra de Fisiopatopatología, hôpital México, UCIMED Y UCR, San José, Costa Rica