cardioprotecció intervinguda per canals de potassi dependents d’ATP.
Dr Fernando Zeledón SA, Dr Orlando Morales MA, Dr Edgar Méndez JB , Dr Eduardo Induni LC, Dr Oswaldo Gutiérrez SD.
Resum
el mitocondri juga un paper central en el manteniment de l’metabolisme de l’cardiomicito durant els fenòmens d’isquèmia i reperfusió. Aquesta “cardioprotecció” sembla estar lligada a l’obertura de canals de potassi dependents d’ATP en la membrana mitocondrial, la qual evita l’obertura de l’porus transicional de permeabilitat (MPTP), la sobrecàrrega de calci i la pèrdua de l’volum de l’espai intermembrana mitocondrial, prevenint la mort cel·lular per necrosi o apoptosi.
Diversos estudis clínics sustenten l’ús prometedor de fàrmacs que obren aquests canals de potassi i que podrien ser una nova arma terapèutica contra la malaltia isquèmica i les seves conseqüències.
Paraules clau: Precondicionament isquèmic; canals de potassi mitocondrials i sarcolémicos; sobrecàrrega de calci; estrès oxidatiu; apoptosi, porus transicional de permeabilitat mitocondrial.
Abstract
Mitochondrial ATP-sensitive potassium channels play an important role preventing necrotic cell death and apoptsis during ischaemia / reperfusió fenomena by pixen of preventing mitochondrial permeability transition pore (MPTP) opening, intracellular calcium overload and loss of mitochondrial intermembrane space.
There is clinical evidence of beneficial effects of a group of drugs called potassium channel Openers that colud be a new therapeutic tool against cardiac ischaemic disease and its consequences.
Keywords: Ischemic preconditioning sarcolemic and mitochondrial potasium channels, calcium overload, oxidative estrès, apoptosi, mitochondrial permeability transitional pore.
Introducció
fins a la data el tractament de la malaltia cardíaca d’origen isquèmic s’ha centrat en prevenir el dany isquèmic, incrementant l’aportació d’oxigen cap a l’àrea miocàrdica en perill o disminuint el consum d’oxigen de l’miocito cardíac.
Igualment, des de fa molts anys enrere se sap que l’exposició de miòcits a breus esdeveniments isquèmics reiteradament, produeix protecció contra posteriors esdeveniments d’isquèmia més duradors, procés anomenat precondicionament isquèmic (1).
Actualment, l’interès científic es concentra en buscar els mecanismes cel·lulars involucrats en la cardioprotecció davant esdeveniments d’isquèmia / reperfusió, entre els quals els canals de potassi sensibles a ATP (K ATP) semblen tenir un paper central (1-3).
en aquest article es revisen conceptes de fisiologia cel·lular i molecular relacionats a el paper dels K ATP en la defensa isquèmica miocardíaca.
Estructura i Funció dels Canals de potassi Cardíacs.
el potassi (K +) és el segon catió més abundant de l’organisme. Un adult de 70 kg conté prop de 4200 mEq de potassi o prop de 50 mEq / kg en l’home i de 40 mEq / kg en la dona, tenint en compte l’ajust per adipocidad i massa corporal. El contingut de K + de l’organisme declina amb l’edat en prop de 2 mEq / kg per cada 10 anys per la disminució de la massa muscular.
Aproximadament, el 98% de K + es troba en el líquid intracel·lular, amb una concentració de 150 mEq / L, mentre que fora de les cèl·lules és de 4 mEq / L, amb un estret rang de normalitat entre 3,5 i 5,5 mEq / L.
La quantitat ingerida de K + en països occidentals és de gairebé 50 a 100 mEq / dia o més o menys 0,7-1,3 mEq / kg de pes corporal per dia. Per exemple, en una persona que consumeix 80 mEq en un dia, el ronyó s’encarregarà d’excretar 70 mEq, el tracte gastrointestinal 9 mEq i la pell al voltant d’1 mEq.
El potassi es troba en una estreta balanç corporal, part de el qual es deu a una redistribució entre els diferents compartiments cel·lulars, ja sigui que aquest ió es traslladi de l’mig intracel·lular a l’extracel·lular o viceversa (4). Perquè tal fenomen passi, la cèl·lula es val de proteïnes transmembrana que funcionen com a canals.
Els canals iònics presenten tres propietats essencials:
1) Un túnel central o porus a través del qual flueixen ions que fa al seu gradient electroquímic.
2) Un filtre de selectivitat, el qual dicta a quin ió se li permetrà el pas a través de l’porus, anomenat regió p
3) Una estructura que exerceix la funció de comporta controlant la probabilitat conformacional d’obertura i tancament de l’canal i per tant, la permeabilitat d’aquesta proteïna (4,5,6,7).
Els canals de potassi estan compostos per subunitats alfa i beta. La responsable de la conducció d’ions a través de la bicapa lipídica és la subunitat alfa.Depenent de la seva topologia i de el nombre de regions P (porus de selectivitat) s’ha establert una classificació dels canals de potassi (4,7,8).
Així per exemple, els canals de potassi que augmenten o disminueixen la seva permeabilitat depenent de l’voltatge transmembrana, presenten sis segments transmembrana (TM) i un porus (1 P) i es denominen canals de K + dependents de voltatge (Kv, figura 1, A); els que presenten 2 TM-1P, són canals que permeten el pas de l’potassi cap al intracel·lular quan la membrana plasmàtica es troba a un voltatge més negatiu que el seu potencial d’equilibri i es troben tancats a potencials més positius, de manera que es denominen, canals de potassi rectificadors cap a dins (Kir, K inwardly rectifyer) (7,9,10,11,12,13).
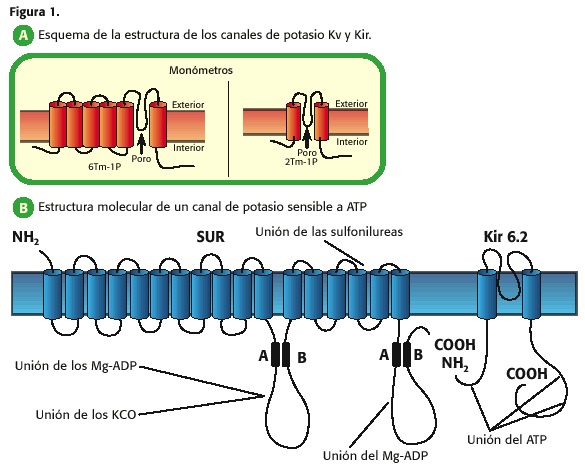
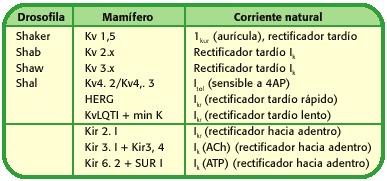
Cadascuna d’aquestes famílies de canals de potassi pot al seu torn presentar subfamílies, depenent de l’electrofisiologia de canal o de l’lligant que hi hagi l’obertura de tals proteïnes, quadre 1.
Quadre 1. Nomenclatura i estructura dels canals de potassi (14).
a Dins dels canals rectificadors cap a dintre es troben dues subfamílies importants en la fisiologia cardíaca: 1) els canals de K regulats per proteïnes G, que són activats pels r eceptores muscarínics M3 (Kach) i 2) els canals de K sensibles a ATP (KATP), aquestes últimes objecte central de la revisió (7,9,10,13).
La funció dels K ATP és més coneguda en les cèl·lules pancreàtiques beta, on s’acoblen canvis en l’excitabilitat elèctrica de la membrana plasmàtica i l’alliberament d’insulina, en relació a la concentració sanguínia de glucosa. No obstant això, també s’han vist involucrats en la protecció neural en els esdeveniments isquèmics i en l’epilèpsia, regulació de el to vascular (per exemple, en la hipertensió arterial pulmonar i sistèmica), recaptura de glucosa en el múscul esquelètic i protecció contra la isquèmia de l’miocito cardíac, tema d’aquesta revisió (10).
Aquestes propietats deriven de l’habilitat dels canals de KATP d’acoblar el metabolisme cel·lular a l’activitat elèctrica, detectant canvis en el citosol dels nivells d’ATP , i d’adenosina difosfat (Mg-ADP), funcionant l’ATP com un bloquejador de canal, i el Mg-ADP com un activador o promotor de la seva obertura (10,11).
el porus dels canals de potassi és un tetràmer i pel fet que els Kir són 2 TM-1P, quatre d’aquestes unitats s’uneixen per formar el porus, denominades a nivell cardíac Kir 6.2 i Kir 6.1. A més, hi ha una altra proteïna anomenada receptor d’sulfonilurea (SUD, sulphonylurea receptor, figura 1, B (15))
que regula l’obertura o tancament de l’porus Kir 6.2 o Kir 6.1: l’ATP inhibeix el canal ja que aquest s’uneix a la subunitat Kir 6, mentre que el Mg-ADP l’activa a través de la seva interacció amb la subunitat SUD (10,11,12,13,14).
Funció central de la mitocòndria en la cardioprotecció
els mitocondris es troben en gairebé totes les cèl·lules amb excepció dels hematies i el seu nombre varia segons el tipus cel·lular; per exemple cada hepatòcit posseeix de 1.022 a 2.000 mitocòndries, que mesuren 3 micres de llarg aproximadament.
Els mitocondris posseeixen dues membranes, una d’externa i una altra interna, que donen lloc als compartiments intermembranosos i a la matriu mitocondrial. A la matriu mitocondrial i en la membrana interna és on es desenvolupen la majoria d’activitats relacionades a la cadena respiratòria (16) .A la mitocòndria ha diverses proteïnes que juguen particulars funcions, ja sigui promovent la cardioprotecció o el detriment de la funció cardíaca; i es troben interrelacionades pels KATP per diferents mecanismes.
Dins d’aquestes les més importants són:
1) El porus transicional de permeabilitat (MPTP, mitochondrial permeability transition pore) que està compost per:
– El canal d’anions dependent de voltatge (VDAC, voltage – dependent anion channel), que se situa en la membrana mitocondrial externa (MME)
– El transportador de nucléotidos d’adenina (ANT, adenine nucleotide translocator), localitzat a la membrana interna (MMI)
– la creatina cinasa (CK), a l’espai intermembranós (EIM)
2) els contra-transportadors Na + / H +, Na + / Ca ++, K + / H +, H + / piruvat, el cotransportador Ca ++ i les proteïnes ja conegudes de la cadena respiratòria, de la beta-oxidació i de l’ cicle de Krebs (17,18) (figura 2).
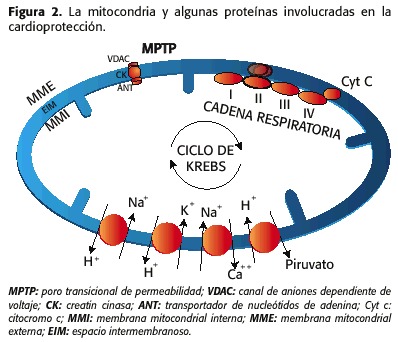
Els esdeveniments més importants involucrats en la gènesi de la lesió per isquèmia / repercussió són:
1. Canvis de l’volum de la matriu mitocondrial
2. L’estat de l’EIM
3. La permeabilitat de la MMI
4. La ruptura de la MME i
5. La sobrecàrrega de calci-sodi intramitocondrial.
Fisiologia mitocondrial de l’ús de l’calci.
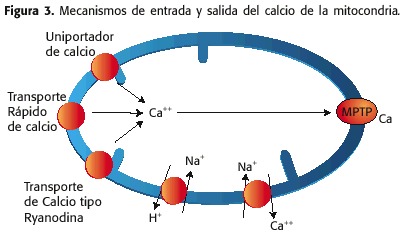
El calci té un paper fonamental en la fisiologia mitocondrial, les concentracions es troben en estrets rangs. El calci pot ser recapturat cap a la matriu mitocòndria a través de la MMI per tres mecanismes:
1) Una bomba de calci, coneguda com el uniportador de calci o UP,
2) un canal que permet l’entrada ràpida de calci, conegut com a proteïna de transport ràpid de calci o RAM (rapid mode Ca ++ uptake) i
3) un receptor tipus rianodinia o RYR (ryanodine receptor).
d’altra banda, la sortida de l’calci de la matriu mitocondrial es deu a:
1) el contra-transportador Na + / Ca ++, en condicions fisiològiques
2) El MPTP, en condicions patològiques (Figura 3)
La funció primordial de l’calci a nivell del mitocondri és l’estimulació de la cadena respiratòria en múltiples nivells, provocant un augment en la producció d’ATP i la seva exportació a l’citosol per satisfer la demanda metabòlica de l’miocito.
També es sap que el procés de fosforilació oxidativa genera una pe tita quantitat de radicals lliures com a producte dels complexos I, coenzim Q i complex III, i que lluny de ser nocius, més aviat són necessaris per a la transducció de senyal de múltiples vies metabòliques intramitocondriales (19).
mecanismes que promouen la sobrecàrrega de calci intramitocondrial. Durant la isquèmia miocàrdica, l’increment de la glucòlisi causa una progressiva acumulació d’àcid làctic, disminuint el pH mitocondrial i que exercirà una retroalimentació negativa que eventualment
s’inhibirà la glucòlisi i la producció d’ATP. L’activació de l’antiportador Na + / H + que s’extruye els hidrogenions del mitocondri procura la normalització de l’pH intramatricial, però en el procés es fomentarà una sobrecàrrega de sodi, el qual no pot ser bombejat fora de la cèl·lula, ja que la Na + / K + -ATPasa es troba inhibida per la pèrdua d’ATP (19). Conseqüentment, l’activitat de l’contra-transportador Na + / Ca ++, el qual usualment exporta calci del mitocondri i de la cèl·lula, es troba disminuïda o fins i tot actuant inversament (a causa de que aquest és un mecanisme de transport actiu secundari) pel que en els diferents compartiments cel·lulars inicia una retroalimentació positiva que promou enormement una sobrecàrrega de calci. En aquest punt, l’homeòstasi iònica no pot ser mantinguda, les concentracions intracel·lulars de sodi i calci progressivament augmenten, amb disminució dels nucleòtids d’adenina i augment de l’fosfat.
A més, l’augment de calci mitocondrial promou la formació de radicals lliures en el mitocondri pels següents mecanismes:
1) la sobrecàrrega de calci mitocondrial promou un augment de l’flux d’electrons en la cadena respiratòria
2) Estimula també la òxid nítric sintetasa, formant òxid nítric (NO)
3) És conegut que el NO inhibeix la cadena respiratòria a nivell de l’complex IV el que
4) Augmenta la formació de radicals lliures ( ROS, reactive oxygen species) pel cicle Q
5) Els complexos i i II de la cadena respiratòria també poden ser inhibits per l’augment de l’calci i NO, contribuint encara més a la formació de ROS
6) el calci també dissocia el citocrom c de la proteïna cardiolipina a la MMI i per darrer mo
7) L’increment de calci promou l’obertura de l’MPTP, permetent l’alliberament de citocrom ca través de la MME (19).
Malgrat tot l’anterior, durant la isquèmia el pH mitocondrial (pHmi) es manté baix i aquest fenomen, com es comentarà en les següents seccions, és prou potent per mantenir el MPTP tancat, sempre que l’esdeveniment d’isquèmia no es prolongui o sobrevingui la tan temuda reperfusió.
El Poro Transicional de Permeabilitat (MPTP)
El MPTP va ser descrit per Haworth i Hunter fa més de 25 anys, però, és fins fa pocs anys que la seva funció ha pres gran importància en els estats d’isquèmia-reperfusió (20,21,22). En un estat estacionari o de homeostasia mitocondrial, el MPTP es troba tancat, la distància
entre la MME i la MMI és òptima i d’aquesta manera hi ha un acoblament entre la VDAC-CK-ANT, el que permet que el principal mecanisme energètic del mitocondri sigui el sistema creatina-fosfocreatina i en segon lloc el d’ATP / ADP (figura 4) (23). A més, la permeabilitat de l’ANT-VDAC és baixa en aquest estat i els canals de potassi dependents d’ATP es troben tancats (18,23,24).

D’altra banda, el antitransportador K + / H + treu potassi de la matriu en intercanvi per un hidrogenión, i també presenten una baixa cinètica en condicions estacionàries, per assegurar així un manteniment de la fosforilació oxidativa, el volum adequat matricial i un potencial de membrana mitocondrial negatiu, ja que com és conegut, el potencial d’hidrogenions entre el EIM i la matriu mitocondrial és mantingut a la impermeabilitat de la MMI als diferents ions, la qual cosa permet la generació d’ATP pel complex V ubicat a la MMI.
en conclusió, des d’un punt de vista fisiològic, la regulació de l’volum de la matriu mitocondrial té importants conseqüències per al metabolisme energètic de l’miocito i l’estat impermeable de el porus contribueix a aquesta homeòstasi (17,24,25).
el poro Transicional de Permeabilitat durant la isquèmia i durant la repercussió. Sota condicions d’isquèmia miocàrdica, l’MPTP s’obre en menor quantia, permetent que la MMI sigui permeable a qualsevol molècula < 1,5 kDa. Es produeixen llavors dues conseqüències importants. Primer, les proteïnes intramitocondriales no poden passar pel porus i exerceixen una pressió coloidosmótica en la matriu amb tumefacció matricial. La MMI per la seva consistència no pateix lisi, però, tal fenomen sí que passa a la MME, amb l’alliberament de proteïnes a l’EIM, com ara el citocrom ci el factor inductor de l’apoptosi que juga un paper crític en la mort cel·lular per aquest mecanisme. Segon, la MMI es torna permeable als protons, el que desacobla la cadena respiratòria, disminució de la producció d’ATP i tercer, es promou el funcionament invers de l’ATP sintetasa, és a dir, en comptes de sintetitzar ATP es promou la hidròlisi de aquest per tal de mantenir un gradient de H + i un potencial de membrana mitocondrial negatiu (18,26,27,28,29).
No obstant això, davant d’aquesta situació, les concentracions d’ATP ràpidament declinen, conduint a una alteració iònica de l’homeòstasi metabòlica ia l’activació d’enzims que promouen la degradació, com ara les fosfolipases, nucleases i proteases. Llevat que passi tancament de l’MPTP, aquests canvis causessin un dany irreversible de la cèl·lula, resultant en necrosi cel·lular.
Un factor clau en l’obertura de l’MPTP és la sobrecàrrega de calci intramatricial, especialment quan aquest s’acompanya d’estrès oxidatiu, depleció de nucléotidos d’adenosina, elevades concentracions de fosfat iònic, i despolarització de la MMI, microambient que es presenta en la isquèmia. Per tant, l’obertura de l’MPTP és un pas crític en la transició de el dany cel·lular reversible a irreversible (18,23).
Com es va esmentar anteriorment, el MPTP es troba format per l’ANT-VDAC i per una altra proteïna que s’anomena ciclofilina-d (Cyp-d), la qual ha de unir-se a l’ANT per provocar la seva obertura, unió que és promoguda per l’augment de la concentració de calci, per la depleció de les reserves de nucléotidos d’adenosina , per l’increment de l’pHmi i alteracions de l’volum matricial. No obstant això, un potent inhibidor de l’obertura de l’porus és la disminució de l’pHmi, fenomen que ocorre durant la isquèmia, establint-se un balanç entre els factors que afavoreixen la seva obertura (sobrecàrrega mitocondrial de calci, depleció d’ATP, augment de l’estrès oxidatiu) i els que el mantenen tancat (sota pHmi), figura 5.
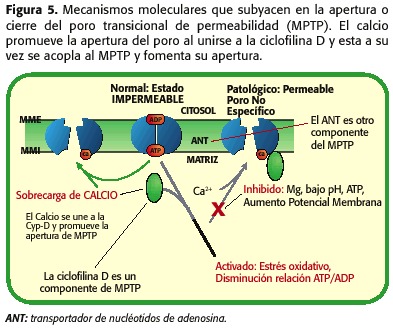
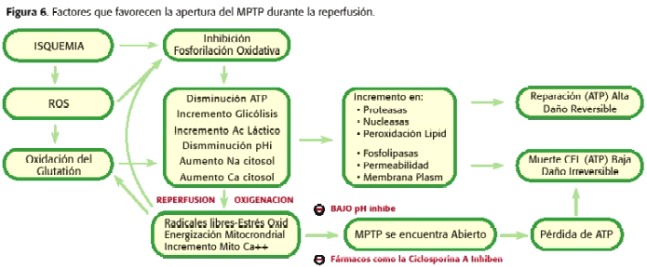
Per tant, durant la isquèmia s’obre el MPTP però en baixa quantitat, ja que el microambient cel·lular no és l’òptim perquè tal fenomen passi. En canvi, durant la reperfusió es produeixen una sèrie de fenòmens intramitocondriales que promouen l’obertura de l’MPTP encara més que durant la isquèmia. Quan ocorre la reperfusió, mitocondri novament és capaç de respirar i generar un potencial de membrana que permeti la síntesi d’ATP, amb l’augment de la producció de radicals lliures com a productes de la cadena respiratòria, el pHmi comença augmentar i encara persisteixen la sobrecàrrega de calci i la depleció d’ATP. Per tant, en aquest punt sí que es promou l’obertura de l’MPTP (30,31,32,33,34,35) (figura 6). Depenent de la proporció de porus oberts i de el temps que romanguin en tal estat propiciarà la necrosi o l’apoptosi (18,23,24) (figura 7).
 a
a 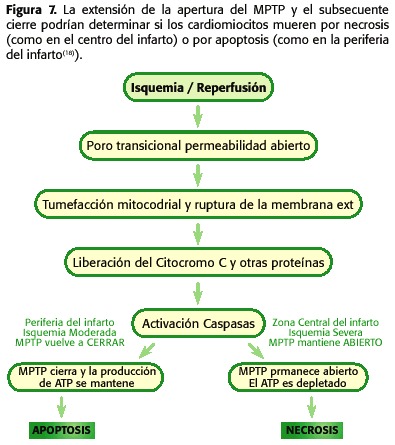
d’aquesta manera, el mitocondri es torna un objectiu terapèutic, ja sigui promovent la inhibició directa de l’MPTP o indirectament, mitjançant la prevenció dels fenòmens que promouen la seva obertura, com ara
1) la sobrecàrrega de calci que es pot aconseguir amb el propofol36 o amb l’augment de les concentracions de Mg ++ que inhibeix a l’contra-transportador Na + / Ca ++ i als canals calci tipus l (37)
2) millorant la bioenergètica mitocondrial o el volum matricial, tots dos fenòmens propiciats amb els fàrmacs que obren els canals de potassi (KCO, potassium channel Openers) veure més endavant
3) mantenint un pH baix durant la reperfusió, per exemple, inhibint el antitransportador Na + / H + amb medicaments com el amiloride, o agregant piruvat (38) a la mitocòndria que fomenta l’augment d’àcid làctic.
Tot el a nterior obre una immensa porta a la recerca en ciències bàsiques i en la terapèutica per a pacients que pateixen d’isquèmia miocàrdica o les seves conseqüències.
Canals de potassi Sensibles a l’ATP: la seva funció en la prevenció de l’obertura de l’MPTP mitocondrial i en la membrana sarcoplásmica
Durant situacions d’estrès miocàrdic, en els quals es depleta l’ATP, els canals de K + augmenten el seu estat d’obertura amb conseqüències beneficioses per al microambient de l’miocito (39,40, 41).
l’obertura dels K ATP mitocondrials causa un modest increment de la influència de K + cap a l’interior de la matriu, el que provoca dos efectes diferents depenent de l’estat bioenergètic de l’cardiomiòcit. En primer lloc, quan la cèl·lula es troba en repòs, el potencial de membrana mitocondrial és alt, i l’entrada d’ions de potassi promou la tumefacció mitocondrial i la alcalinització de la matriu, el que fomenta un lleuger increment en la producció de radicals lliures . En segon lloc, si la cèl·lula es troba en estat d’isquèmia, mitocondri es troba despolaritzada (potencial baix), el que promou la sortida de K +, fenomen que és contrarestat pels canals de K ATP i per tant es prevé la contracció de l’ volum matricial que d’una altra manera passaria (42).
l’ingrés de potassi a la matriu mitocondrial genera una producció lleugera de radicals lliures (ROS), i com està demostrat, aquests juguen un important paper com a segons missatgers en una varietat de senyals intracel·lulars (39). Aquesta producció de ROS en una cèl·lula en repòs a més promou la recaptura de K + en intercanvi per un H + (antitransportador K + / H +), el que crea el gradient per a l’intercanvi d’un fosfat (Pi) pel cotransportador electroneutro Pi / H +. La recaptura de Pi és molt menor que la de K +, a causa que el Pi està present en molt menor concentració que el K +. Per aquesta raó, el pH de la matriu sempre s’incrementa quan el volum de la matriu també augmenta, fenòmens promoguts per la recaptura de Pi i K + (23).
Quan encara no han entrat en funció dels canals de KATP durant esdeveniments isquèmics, la matriu mitocondrial pateix una contracció i el EIM augmenta, amb la desunió de l’VDAC de l’ANT pel CK, la qual cosa augmenta la conductància a l’intercanviador de nucleòtids pel VDAC i l’ANT, contrari al que passa a repòs, contribuint així a la depleció d’ATP matricial i citosòlic (figura 3). Per tant, l’obertura dels KATP durant la isquèmia, per exemple promoguda pel diazòxid, manté el volum de l’EIM, redueixen la taxa de pèrdua d’ATP, redueix la taxa de la degradació dels nucleòtids d’adenina de tal manera que existiran reserves d’ADP per a la posterior fosforilació durant la reperfusió i finalment, redueix els canvis en el potencial de membrana mitocondrial i l’acumulació de Ca ++, prevenint la sobrecàrrega de calci, ja que l’ATP es manté en concentracions adequades per al funcionament mínim de la Na + / K + ATPasa i altres bombes.
Aquests efectes preserven la funció mitocondrial i d’aquesta manera es pot fer front a la reperfusió amb millors resultats fisiopatològics. Durant la reperfusió, la subseqüent obertura de canals de KATP permet la compartimentalització dels nucleòtids d’adenina, el que significa que la primera font d’energia és el fosfat provinent de la fosfocreatina i no de l’ATP (baixa conductància de l’VDAC (23), figura 3).
per tant, l’activació dels canals de K ATP mitocondrials durant la isquèmia-reperfusió prevé la formació de l’porus transicional de permeabilitat inhibint l’estrès oxidatiu, desencadenat per l’ingrés de Ca ++ ( 18,23,24).
Vanden Hoek et al. van demostrar que l’augment de radicals lliures en el mitocondri durant el precondicionament isquèmic era necessari per protegir el miocito contra un subseqüent estrès oxidatiu durant la reperfusió.És així com s’ha hipotetitzat que hi ha dues etapes en l’alliberament de radicals lliures, la primera que contribueix a l’obertura dels canals de potassi sensibles a l’ATP i una posterior durant la repercussió, que és nociva i desencadena remodelat i apoptosi cardíaca (42 ). A la membrana sarcolémica de l’miocito cardíac i del múscul llis, l’obertura dels canals de potassi en la també promou efectes protectors.
Així, és beneficiosa la hiperpolarització cel·lular que prevé la sobrecàrrega de Ca ++ intracel·lular, disminuint la durada de l’potencial d’acció, limitant el dany cel·lular i preservant les reserves energètiques cel·lulars i per tant, la supervivència de l’miocito. A més, aquesta seqüència pot ser tant aguda com crònica figura (8,42,44). A nivell vascular l’obertura d’aquests canals promou vasodilatació, deguda a la hiperpolarització cel·lular ia la reducció en l’entrada de Ca ++, amb augment de l’flux coronari i disminució de la postcàrrega.
Perspectives Clíniques
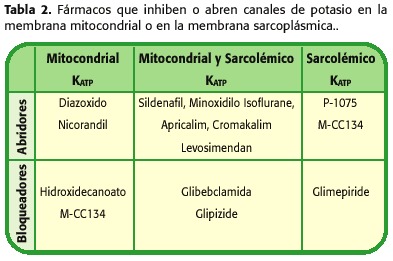
a causa de les propietats combinades de cardioprotecció i vasodilatació, els medicaments que permeten l’obertura dels canals de potassi (KCO), podrien ser considerats per a determinades condicions cardíaques (quadre 2). Aquestes inclouen protecció de l’miocardi sota circulació extracorpòria, preservació de cor donant en el trasplantament cardíac, tractament de la malaltia isquèmica cardíaca, de la hipertensió arterial sistèmica i pulmonar, malaltia vascular perifèrica i en les arítmies relacionades amb una repolarització anormal (41-50) .
 a
a
En cirurgia cardíaca, els KCO podrien jugar un paper important a les solucions de cardioplegia. En diversos models de cirurgia cardíaca amb circulació extracorpòria, diversos KOC incloent nicorandil, aprikalim i pinacidil, van promoure major cardioprotecció que amb cardioplegia convencional (51,52).
En pacients als quals se’ls va realitzar cirurgia de ponts (bypass) coronaris, el temps requerit per assolir l’arrest cardíac, els canvis en el segment ST després de l’pinçament aòrtic, els nivells plasmàtics de la CPK-MB i dosi d’agents inotrópicos, van ser tots menors que el grup tractat amb nicorandil comparat amb els controls tractats amb teràpia convencional (53).
la utilització de solucions de cardioplegia amb KCO com ara el diazòxid, el minoxidil (54), el propofol (que inhibeixen la sobrecàrrega de calci ++ (36)) , el magnesi (que inhibeix la sobrecàrrega de calcio37), els digitàlics (que fomenten cardioprotecció per vies de senyalització cel·lular que convergeixen amb les dels KCO (23,55)), el piruvat (que fomenta la acidosi intracel·lular i normalització gradual de l’pH pel tancament de l’MPTP (38)), els inhibidors de l’contra-transportador Na + / H ++ com ara el amiloride, (que eviten també la pèrdua de l’efecte protector de l’PHI baix), l’adenosina (que també és un KCO, actuant mitjançant proteïnes G, prevé la sobrecàrrega de calci ja que la cardioplegia hiperpotasémica la promou (52,56)), poden ser forts objectius per a la investigació en la preservació de la funció cardíaca en cirurgies d’alt risc i poden evitar complicacions potencialment mortals com la síndrome de baixa despesa post-bomba i moltes altres, amb disminució dels costos econòmics a l’reduir les estades hospitalàries.
d’altra banda, l’ús d’antioxidants semblés ser beneficiós només en determinades circumstàncies, ja que el seu ús en assajos de laboratori ha bloquejat el precondicionament isquèmic, a causa que s’alliberen radicals lliures en menor quantia (23,25).
en la síndrome coronària aguda, el valor dels KCO està millor documentat clínicament amb nicorandil que ha demostrat ser beneficiós amb mínims efectes adversos en el maneig tant de l’angina estable com inestable (57). En un estudi multicèntric que va involucrar més de 5.000 pacients amb angor estable, l’ús a llarg termini de nicorandil es va associar amb reducció en els esdeveniments cardiovasculars com ara mort cardíaca, infart miocàrdic i hospitalització a causa de dolor toràcic (58,59).
En pacients amb angor inestable, el nicorandil sumat a un agressiu tractament anti-isquèmic va reduir els episodis d’isquèmia / necrosi miocàrdica i arítmies quan es va comparar amb pacients tractats amb la teràpia convencional. En pacients sotmesos a angioplastía coronària, el nicorandil precondicionó el cor, va millorar l’hemodinàmica coronària i va preservar la viabilitat miocàrdica davant la reperfusió (60,61,62).
El nicorandil també disminueix la precàrrega i la postcàrrega, augmenta l’alliberament d’òxid nítric per les cèl·lules endotelials i, a diferència de la nitroglicerina, no desenvolupa tolerància cap als seus efectes antianginosos (63).
En l’angina vasoespástica, el nicorandil és un potent vasodilatador i s’ha demostrat que disminueix els episodis de l’angina variant, disminueix els canvis de l’segment ST i millora la perfusió coronària. Els KOC també han demostrat millors resultats en la cirurgia de bypass coronari utilitzant empelts arterials (artèries toràcica interna, gastroepiploica, radial) ja que evita el vasoespasme posterior a la cirurgia (64,65,66)). Es requereix més experiència i dades científiques amb l’ús d’KCO en aquestes situacions clíniques.
L’activació dels KATP sarcolémicos és responsable del corrent elèctric que subjau a l’elevació de l’segment ST, l’indicador electrocardiogràfic clàssic de dany isquèmic transmural miocàrdic (41). Els pacients amb diabetis mellitus tractats amb sulfonilurees i que estan cursant un infart agut a el miocardi, presenten una magnitud atenuada en l’elevació de l’ST, la qual cosa dificulta el diagnòstic inicial, ja que les sulfonilurees són inhibidors dels canals de KATP dependents (41) .
Conclusió
a l’igual que pràcticament totes les malalties humanes, la terapèutica de la malaltia isquèmica cardíaca es fonamenta en els nous coneixements en la fisiologia i fisiopatologia cel·lular. La modulació dels canals KATP és un procés crític en l’homeòstasi metabòlica de la cèl·lula i conforme la investigació biomèdica dilucidi nous conceptes sobre l’estructura, funció, regulació i selectivitat tissular d’aquests canals, nous agents terapèutics es podran desenvolupar amb enormes avantatges per a la població en risc.
Referències
1. Ardehali H, O Rourke. Mitochondrial KATP channels in cell survival and death. J Mol Cell Cardiol 2005; 39: 7-16.
2. Kloner RA, Bolli R, Marban I, Reinilib L, Braunwald E. Medical and cellular Implications of Stunning, hibernation and preconditioning: an NHLBI workshop. Circulation 1998; 97: 18: 48-67.
3. Murray CE, Jennings RB, Reiner KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986; 74: 11: 24-36.
4. Cogan MG. Líquids i electròlits: Fisiologia i Fisiopatologia. Manual Modern. Mèxic, 1993; 145-168.
5. Sansom MSP, Shrivastava IH i at. Potassium channels: structures, models, simulations. Biochimica et Biophysica Acta 2002, 1565: 294-307.
6. Biggin PC, Roosild T, Choe S. Potassium channel structure: domain by domain. Current Opinion Structural Biology 2000, 10: 456-461.
7. Giblin JP, Leaney JL, and Tinker A. The molecular assembly of ATP-sensitive potassium channels: determinants on the pore forming subunit. J Biol Chem 1999, 274: 22.652-22.659.
8. Choe S. Potassium channel structures. Nat Rev Neuroscience 2002, 3: 115-121.
9. Loussouarn G, Rose T, Nichols CG. Structural Basis of Inward Rectifying Potassium Channel Gating. Trends cardiovasc Med 2002; 12: 253-258
10. Nishida M, Mackinnon R. Structural Basis of Inward Rectification: Cytoplasmic Pore of the G Protein-Gated Inward Rectifier GIRK1 at 1,8 A Resolution. Cell 2002; 111: 957-965.
11. Yamada M, Inanobe A, Kurachi I. G Protein Regulation of Potassium Ion Channels. Pharmacological Reviews 1998; 50: 724-747.
12. Bichet D, Haass F, Yeh Jan L. merging Functional Studies with Structures of Inward Rectifier K + Channels. Nature Reviews 2003; 4: 957-67.
13. Aguilar-Bryan L, Clement IV J, González G, Kunjilwar K, Babenko A, Bryan J. Toward Understanding the Assembly and Structure of KATP Channels. Physiological Reviews 1998; 78: 227-242.
14. Elizari M, Chiale P. Arítmies Cardíaques: Fonaments cel·lulars i moleculars, diagnòstic i tractament. Panamericana, Buenos Aires, 2003; 31-40.
15. Campbell JD, Sanson MSP, Ashcroft F. Potassium Channel Regulation. Nature Reviews 2003; 11: 1038-1042.
16. Méndez JE, Zeledón SF, Zamora JF, Cortés VA. Un Acostament a la Cinètica de l’Oxigen Part 1. Rev Costarr Caridiol 2004; 6: 27-32.
17. Suleiman M, Halestrap AP, Griffiths E.J. Mitocondri: a target for Myocardial protection. Pharmacology and Therapeutics 2001; 89: 29-46.
18. Halestrap AP, Clarke J, Sabzali A, Javadov A. Mitochondrial permeability transition pore opening during Myocardial reperfusió: a target for cardioprotection. Cardiovasc Res 2004; 61: 372-385.
19. Brookes P, Yoon I, Robotham J et al. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 2004; 287: C817-C833.
20. Hunter DR and Haworth RA. The Ca ++ induced membrane transition in mitocondri I. The protective mechanisms. Arch Biochem Biophys 1979; 195: 453-459.
21. Hunter DR and Haworth RA. The Ca ++ induced membrane transition in mitocondri II. Nature of the Ca ++ trigger site. Arch Biochem Biophys 1979; 195: 460-67.
22. Hunter DR and Haworth RA. The Ca ++ induced membrane transition in mitocòndria III.Reunió de transició ca ++. Arc Biochem Biophys 1979; 195: 468- 77.
23. Garlid KD, dos Santos P, XIE ZJ, Costa A, Pracek P. Mitocondrial Mitocondrial Transport de potassi: el paper del canal K + Sensible a l’ATP mitocondrial en funció cardíaca i cardioprotecció. Biochem Biophys Acta 2003; 1606: 1-21.
24. Macfalles E, Liem d et al. Funció mitocondrial: el cor de la preservació del miocardi. J Lab Clin Med 2003; 142: 141-9.
25. Facundo H, Fornazari M, Kowaltowski A. Protecció de teixits mediada per canals mitocondrials K +. Biochem Biophys Acta 2005; 1701: 1-11.
26. Halestrap ap. La transició de permeabilitat mitocondrial: el seu mecanisme molecular i el seu paper en lesions de reperfusió. A: Brown GC, Nicholls DG, Cooper CE, editors. Mitocòndries i mort cel·lular. Simposia de la societat bioquímica. Londres: Portland Press; 1999, vol. 66, pàg. 181-203.
27. Halestrap AP, McStay GP, Clarke SJ. El complex de porus de transició permeabilitat: una altra vista. Biochimie 2002; 84: 153-66.
28. Halestrap AP, Kerr PM, Javadov S, Woodfield Ky. Elocidant el mecanisme molecular del porus de transició de la permeabilitat i el seu paper en lesions de reperfusió del cor. Biochim Biophys Acta 1998; 1366: 79-94.
29. Halestrap AP, Brenner C. La tradocasa adenina nucleòtide: un component central del porus de transició de la permeabilitat mitocondrial i el jugador clau en la mort cel·lular. Curr Med Chem 2003; 10: 1507-25.
30. Duchen Sr, McGuinness O, Brown La, Crompton M. Sobre la participació d’una ciclosporina-un porus mitocondrial sensible en lesions de reperfusió de miocardi. Cardiovasc Res 1993; 27: 1790-4.
31. Lemasters JJ, Nieminen Al, Qian T, Trost LC, Herman B. La transició mitocondrial de la permeabilitat en lesions tòxiques, hipòxiques i de reperfusió. MOL Cell BioChem 1997; 174: 159-65.
32. Lemasters JJ, Trollinger Dr, Qian T, Cascio Nosaltres, Ohata H. Imatge confocal de Ca2 +, pH, potencial elèctric i permeabilitat de la membrana en cèl·lules vives individuals. Mètodes enzimol 1999; 302: 341-58.
33. Xu mf, wang yg, hirai k, ayub a, ashraf A. precondicionament de calci inhibeix la transició de la permeabilitat mitocondrial i l’apoptosi. Am j fisiol 2001; 280: H899- 908.
34. Miyata H, Lakatta, per exemple, Stern MD, Silverman Hs. Relació de calci mitocondrial i citosòlic a la recuperació de miocítes cardíaca després de l’exposició a Anoxia. Circ Res 1992; 71: 605-13.
35. Griffiths EJ, Ocampo CJ, Savage JS, et al. Camins de transport de calci mitocondrial durant la hipòxia i la reoxigenació en cardiomiites úniques de rata. Cardiovasc Res 1998; 39: 423-33.
36. Lim khh, modi p, nicholson e, et al. Un model de porc d’arrest cardiopològic de sang calenta per investigar els efectes cardioprotectors de propofol. J Fisiol 2001; 536p: 82p.
37. Headrick JP, McKirdy JC, Willis RJ. Efectes funcionals i metabòlics del magnesi extracel·lular en miocardi normòxic i isquèmic. AM J Fisiol 1998; 275: H917-29.
38. Mallet RT. Piruvat: Protector metabòlic de rendiment cardíac. Proc SoC Exp Exp Biol Med 2000; 223: 136- 48.
39. Oldenburg O, Cohen M et al. Canals katp mitocondrials: paper en cardioprotecció. Cardiovasc Res 55: 429-437, 2002.
40. Minners J, McLeod C, sac N. Plasticitat mitocondrial en precondicionament isquèmic clàssic que es mou més enllà del canal katp mitocondrial. Cardiovas Res 2003; 59: 1-6.
41. Kane G, Liu X, Yamada S, Olson T, Terzic A. Canals de KATP cardíacs en salut i malaltia. J Mol mol Cardiol 2005; 38: 937-943.
42. Wang y, Haider H, Ahmad N, Aschraf M. Mecanismes per que els obridors de canals KATP produeixen cardioprotecció aguda i retardada. Farmacologia vascular 2005; 42: 253-264.
43. Vanden Hoeck t, Becker L.B, Shao Z, Schumacker P. Espècies d’oxigen reactiva alliberades de mitocòndries durant la breu hipoxia indueixen precondicionament en cardiomiòcits. J Biol. Quim de química. 1998; 273: 18092-98
44. Jahangir A, Terzic A. Katp Canal Therapeutics al costat del llit. J Mol mol Cardiol 2005; 39: 99-112.
45. Kane GC, Behfar A, Yamada S, Pérez-Terzic C, O’Cochlain F, et al. Hoenicke em, Sun XW, RG Strange, Damiano RJ. Preservació del cor donant amb una nova solució hiperpolaritzadora: protecció superior en comparació amb la solució de la Universitat de Wisconsin. J Thorac Cardiovasc Surg 2000; 120: 746-54.
46. Quast u, Guillon JM, Cavero I. Farmacologia cel·lular d’obertures de canals de potassi en múscul llis vascular. Cardiovasc Res 1994; 28: 805-10.
47. Okada Y, Yanagisawa t, Taira N. BRL 38227 (Levcromakalim) -Inducolarització redueix la sensibilitat a CA2 + d’elements contractils en l’artèria coronària canina. Naunyn Schmiedebergs Arch Farmacol 1993; 347: 438-44.
48. Donnelly R, Elliott Hl, Meredith PA, Reid Jl. Estudis clínics amb l’activador del canal de potassi Cromakalim en temes normotens i hipertensos. J Cardiovasc Farmacol 1990; 16: 790-5.
49. Simpson D, Wellington K.Nicoràndia: una revisió del seu ús en la gestió de l’angina estable Pectoris, incloent pacients d’alt risc. Drogues 2004; 64: 1941-55.
50. McCully JD, Levitsky S. Mitocondrials canals de potassi sensibles a ATP en cardioprotecció quirúrgica. Arc Biochem Biophys 2003; 420: 237-45.
51. Kevelaitis e, Oubenaissa A, Peynet J, MOUAS C, MENASCHE P. Precondicionament per obertures de canals de potassi sensibles a ATP mitocondrials: un enfocament efectiu per millorar la preservació dels trasplantaments cardíacs. Circulació 1999; 100: 345- 50.
52. Steensrud T, Nordhaug D, Husnes KV, Aghaxani E, Sorlie DG. Substitució de potassi amb nicoràndia en fred Hospital de Sant Tomàs de l’Hospital Cardiopegia millora la conservació de l’energia i la funció en cors de porcs. Ann Thorac Surg 2004; 77: 1391-7.
53. Hayashi y, Sawa Y, Ohtake S, Nishimura M, Ichikawa H, Matsuda H Controlada Administració de Nicorandil Protecció Formyocardial durant l’art de l’artèria coronària Blofting sota el bypass cardiopulmonar. J Cardiovasc Farmacol 2001; 38: 21-8.
54. Garlid PD, va treure p i al. Efecte cardioprotector de diazòxid i la seva interacció amb els canals Mitocondrials de K + Sensible: Possible mecanisme o cardioprotecció, Circ Res 1997; 81: 1072-182.
55. Xic Z. Ouabain Interacció amb Cardiac Na / K- Atpasa revela que l’enzim pot actuar com a bomba i com a transductor de senyal. Cell Mol Biol 2001; 47: 383-390.
56. Jovanovic A, López J, Alekseev A et al. L’adenosina evita la càrrega CA21 induïda per K1: Insight de cardioprotecció durant la cardiopia. Ann Thorac Surg 1998; 65: 586-91.
57. Simpson D, Wellington K. Nicorandil: una revisió del seu ús en la gestió de l’angina estable Pectoris, incloent pacients d’alt risc. Drogues 2004; 64: 1941-55.
58. Markham A, Plosker GL, Goa Kl. Nicorandil: una revisió actualitzada del seu ús en malalties del cor isquèmic amb èmfasi en els seus efectes cardioprotectors. Drogues 2000; 60: 955-74.
59. El grup d’estudi IONA. Prova per mostrar l’impacte de Nicorandil a Angina (IONA): disseny, metodologia i gestió. Cor 2001; 85: E9.
60. Ito H, Taniyama Y, Iwakura K, Nishikawa N, Masuyama T, et al. La nicoràndia intravenosa pot preservar la integritat de microvascular i la viabilitat de miocardi en pacients amb un infart de miocardi de paret anterior reperfús. J am Coll Cardiol 1999; 33: 654-60.
61. Schlepper M, Thormann J, Berwing K, Strasser R, Mitrovicv. Efectes de la nicoràndia sobre la perfusió regional i la funció ventricular esquerra. Drogues Cardiovasc Ther 1995; 9: 203-11.
62. Sakatay, Kodama K, Komamura K, Limyj, Ishikura F, et al. EFECTE SALUTARI DE L’ADMINISTRACIÓ NICORANDIL INTRACORONARI ADJUNTIVA SOBRE LA RESTAURACIÓ DEL FLUX SANIA MIOSCARDIAL I MILLORA FUNCIONAL EN ELS PACIENTS AMB INFARCTCK MYOCARDIAL AGUT. CORT J 1997; 133: 616-21.
63. Simpson D, Wellington K. Nicorandil: una revisió del seu ús en la gestió de l’angina estable Pectoris, incloent pacients d’alt risc. Drogues 2004; 64: 1941-55.
64. Kaski JC. Gestió de la angina vasospàstica-paper de Nicorandil. Drogues Cardiovasc Ther 1995; 9: 221-7.
65. Chen JW, Lee WL, Hsu NW, Lin SJ, Ting CT, Wang Sp, et al. Efectes del tractament a curt termini de la nicoràndia sobre l’isquèmia de miocardi induïda per l’exercici i l’activitat autonòmica cardíaca anormal en angina microvascular. Am J Cardiol 1997; 80: 32-8.
66. Akar f, Uydes-Dogan BS, Tufan H, Aslasi s, Koksoy C, Kanzik I. La comparació de la capacitat de resposta de les artèries mamàries internes i gastromàpis de la capacitat d’aïllament humà a Levcromakalim: un enfocament alternatiu a la gestió de l’espasme d’empelt. BR J CLIN Farmacol 1997; 44: 49-56.
Un Departament de Fisiología, Universitat de Ciències Médicas (Ucimed), Sabana Oeste, San José, Costa Rica. Teléfono (506) 296-3944, e-mail: [email protected], [email protected]
B Cátedra de Cirugía, Hospital México, Ucimed, San José, Costa Rica.
C Cátedra de Cirugía, Hospital Mèxic, Universitat de Costa Rica (UCR), San José, Costa Rica.
D Cátedra de Fisiopatología, Hospital México, Ucimed y UCR, San José, Costa Rica